

Loss of G(1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1
- PMID: 10799578
- PMCID: PMC110856
- DOI: 10.1128/jvi.74.11.5040-5052.2000
Loss of G(1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1
Abstract
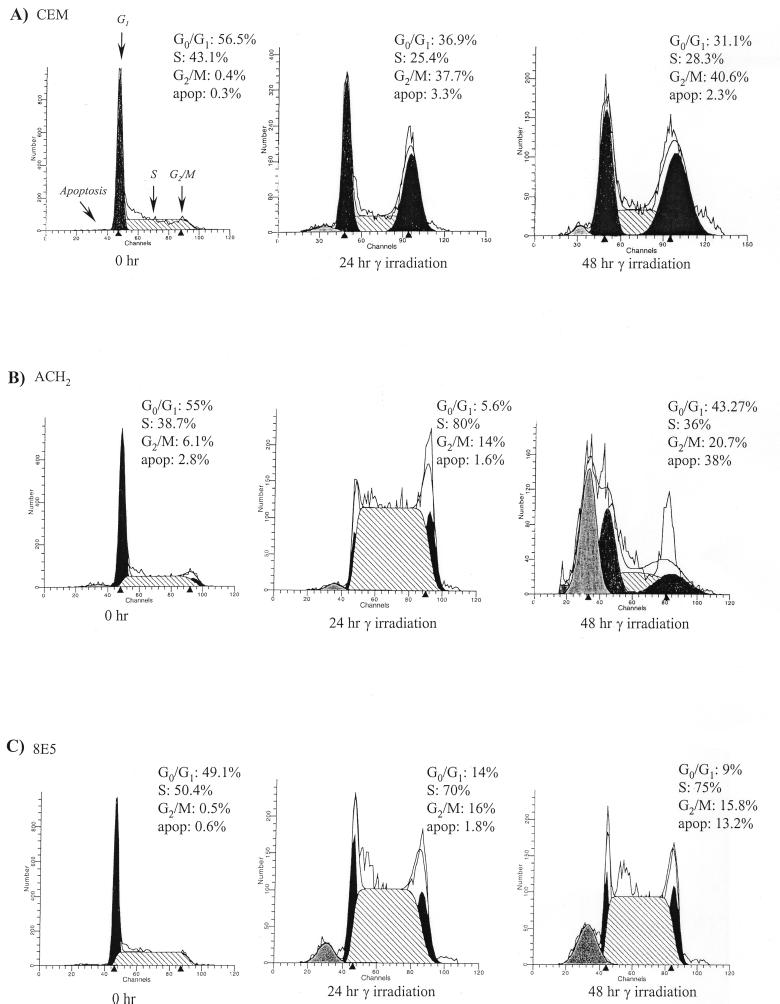
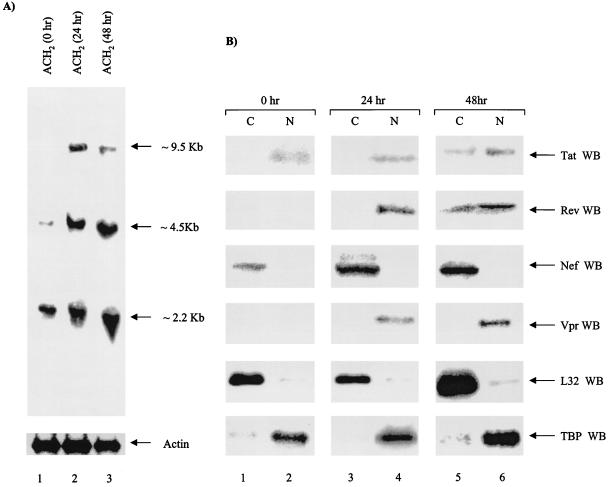
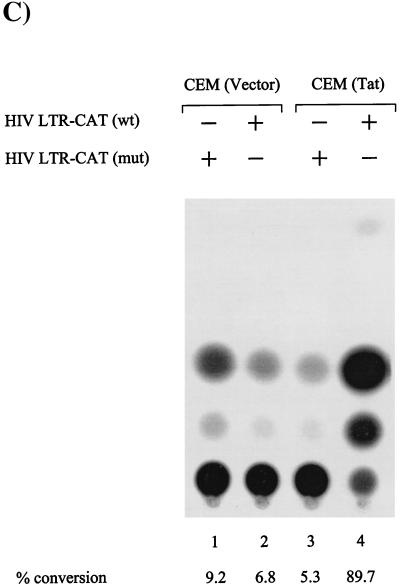
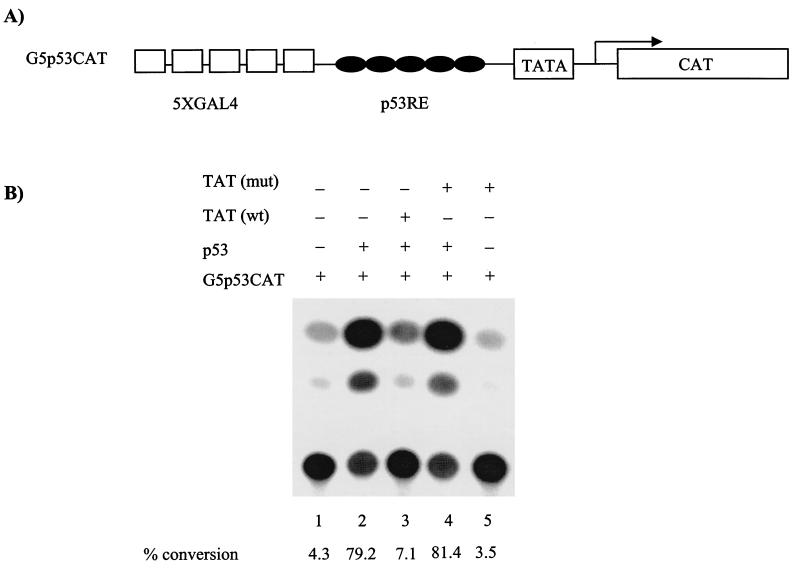
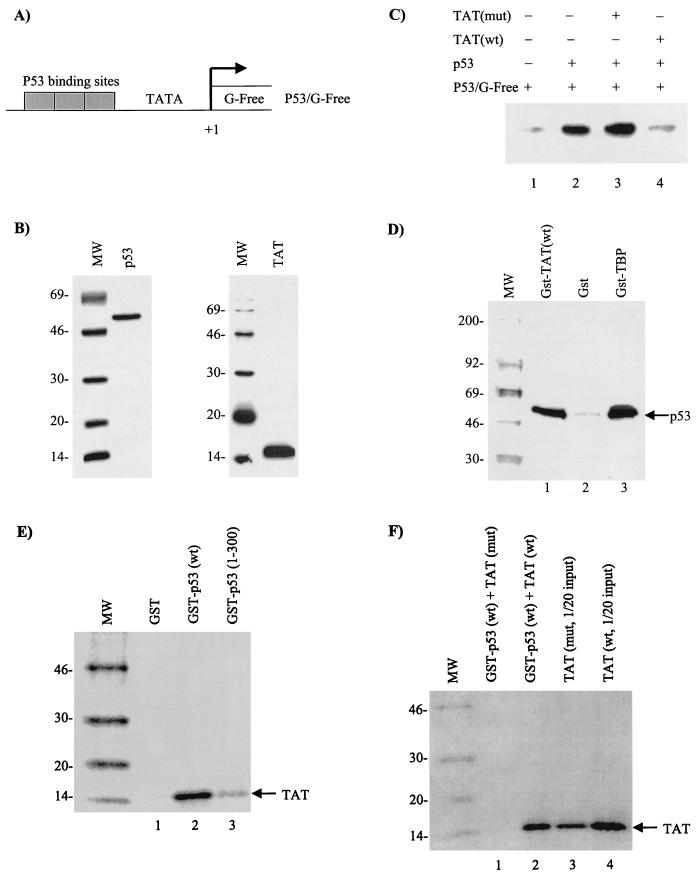
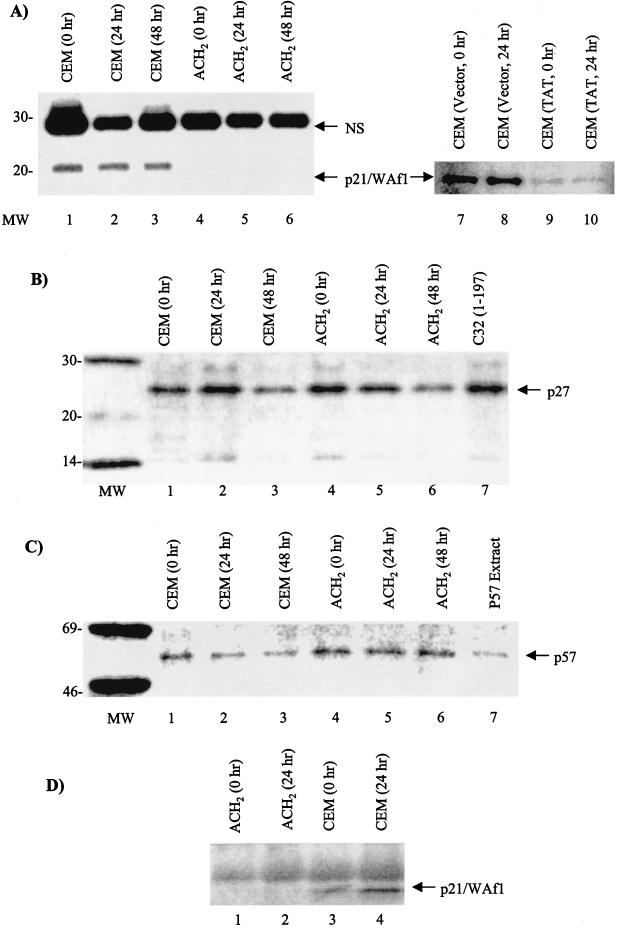
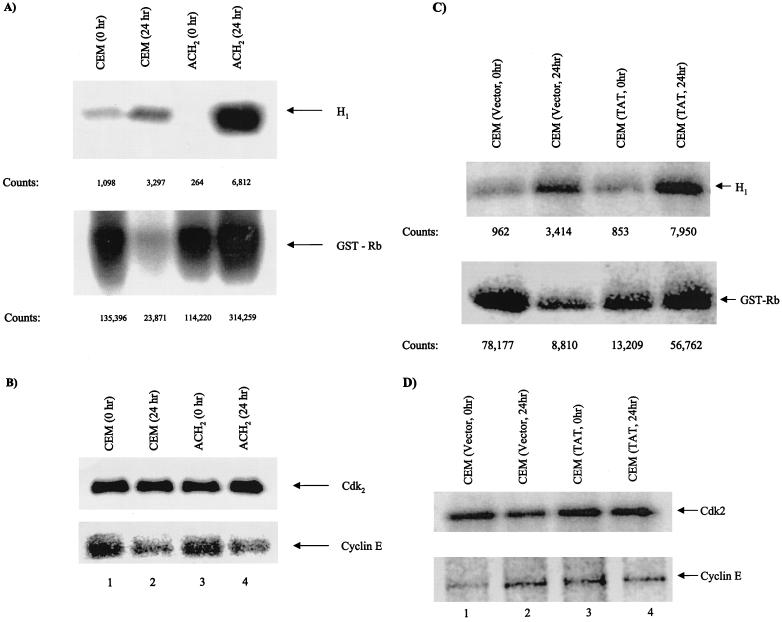
Productive high-titer infection by human immunodeficiency virus type 1 (HIV-1) requires the activation of target cells. Infection of quiescent peripheral CD4 lymphocytes by HIV-1 results in incomplete, labile reverse transcripts and lack of viral progeny formation. An interplay between Tat and p53 has previously been reported, where Tat inhibited the transcription of the p53 gene, which may aid in the development of AIDS-related malignancies, and p53 expression inhibited HIV-1 long terminal repeat transcription. Here, by using a well-defined and -characterized stress signal, gamma irradiation, we find that upon gamma irradiation, HIV-1-infected cells lose their G(1)/S checkpoints, enter the S phase inappropriately, and eventually apoptose. The loss of the G(1)/S checkpoint is associated with a loss of p21/Waf1 protein and increased activity of a major G(1)/S kinase, namely, cyclin E/cdk2. The p21/Waf1 protein, a known cyclin-dependent kinase inhibitor, interacts with the cdk2/cyclin E complex and inhibits progression of cells into S phase. We find that loss of the G(1)/S checkpoint in HIV-1-infected cells may in part be due to Tat's ability to bind p53 (a known activator of the p21/Waf1 promoter) and sequester its transactivation activity, as seen in both in vivo and in vitro transcription assays. The loss of p21/Waf1 in HIV-1-infected cells was specific to p21/Waf1 and did not occur with other KIP family members, such as p27 (KIP1) and p57 (KIP2). Finally, the advantage of a loss of the G(1)/S checkpoint for HIV-1 per se may be that it pushes the host cell into the S phase, which may then allow subsequent virus-associated processes, such as RNA splicing, transport, translation, and packaging of virion-specific genes, to occur.
Figures


Similar articles
-
TGF-beta-mediated cell cycle arrest of HPV16-immortalized human ectocervical cells correlates with decreased E6/E7 mRNA and increased p53 and p21(WAF-1) expression.Exp Cell Res. 2000 Aug 25;259(1):149-57. doi: 10.1006/excr.2000.4953. Exp Cell Res. 2000. PMID: 10942587
-
Overexpression of p21(waf1) in human T-cell lymphotropic virus type 1-infected cells and its association with cyclin A/cdk2.J Virol. 2000 Aug;74(16):7270-83. doi: 10.1128/jvi.74.16.7270-7283.2000. J Virol. 2000. PMID: 10906181 Free PMC article.
-
Cell cycle exit during terminal erythroid differentiation is associated with accumulation of p27(Kip1) and inactivation of cdk2 kinase.Blood. 2000 Oct 15;96(8):2746-54. Blood. 2000. PMID: 11023508
-
G1 phase accumulation induced by UCN-01 is associated with dephosphorylation of Rb and CDK2 proteins as well as induction of CDK inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carcinoma A431 cells.Cancer Res. 1997 Apr 15;57(8):1495-501. Cancer Res. 1997. PMID: 9108451
-
Role of p27(Kip1) in human intestinal cell differentiation.Gastroenterology. 2001 Feb;120(2):423-38. doi: 10.1053/gast.2001.21199. Gastroenterology. 2001. PMID: 11159883
Cited by
-
HIV-1 Tat Recruits HDM2 E3 Ligase To Target IRF-1 for Ubiquitination and Proteasomal Degradation.mBio. 2016 Oct 18;7(5):e01528-16. doi: 10.1128/mBio.01528-16. mBio. 2016. PMID: 27795392 Free PMC article.
-
Hematopoietic Stem and Immune Cells in Chronic HIV Infection.Stem Cells Int. 2015;2015:148064. doi: 10.1155/2015/148064. Epub 2015 Aug 2. Stem Cells Int. 2015. PMID: 26300920 Free PMC article. Review.
-
Therapeutic doses of irradiation activate viral transcription and induce apoptosis in HIV-1 infected cells.Virology. 2015 Nov;485:1-15. doi: 10.1016/j.virol.2015.06.021. Epub 2015 Jul 14. Virology. 2015. PMID: 26184775 Free PMC article.
-
Viral Modulation of the DNA Damage Response and Innate Immunity: Two Sides of the Same Coin.J Mol Biol. 2022 Mar 30;434(6):167327. doi: 10.1016/j.jmb.2021.167327. Epub 2021 Oct 22. J Mol Biol. 2022. PMID: 34695379 Free PMC article. Review.
-
9-Aminoacridine inhibition of HIV-1 Tat dependent transcription.Virol J. 2009 Jul 24;6:114. doi: 10.1186/1743-422X-6-114. Virol J. 2009. PMID: 19630958 Free PMC article.
References
-
- Biggs J R, Kraft A S. Inhibitors of cyclin-dependent kinase and cancer. J Mol Med. 1995;73:509–514. - PubMed
-
- Brugarolas J, Chandrasekaran C, Gordon J I, Beach D, Jacks T, Hannon G J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. - PubMed
-
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown J P, Sedivy J M, Kinzler K W, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
