

BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M
- PMID: 10567572
- PMCID: PMC84954
- DOI: 10.1128/MCB.19.12.8469
BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M
Abstract
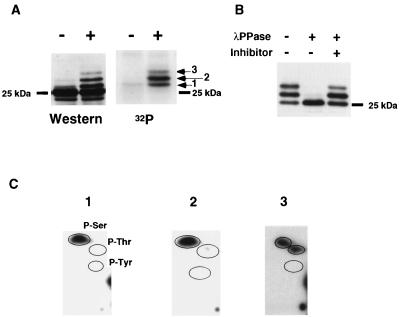
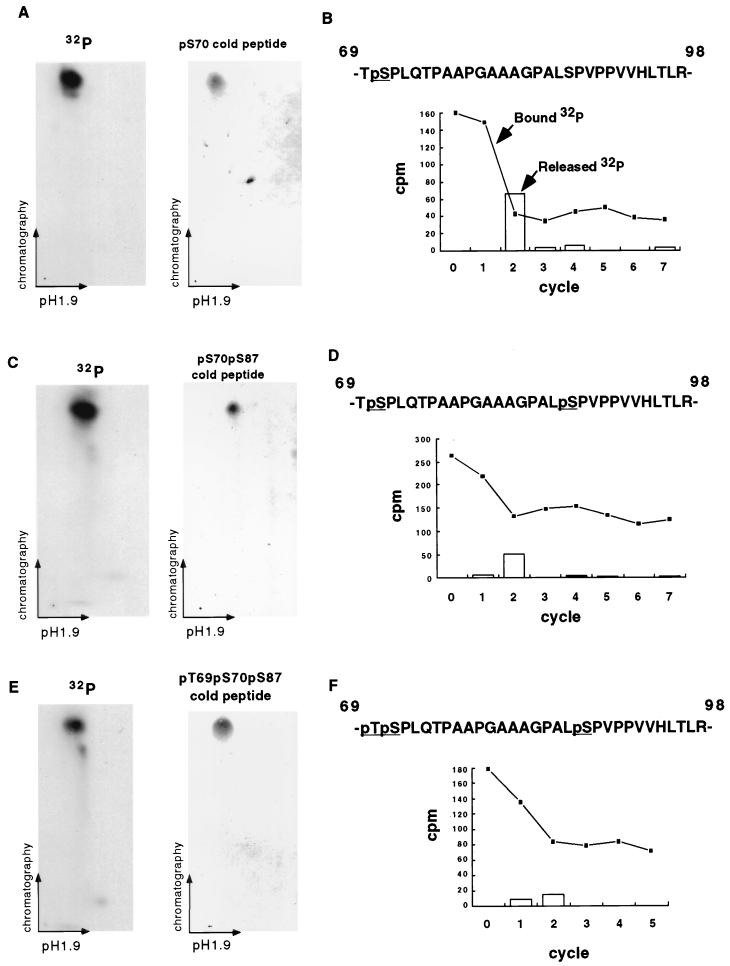
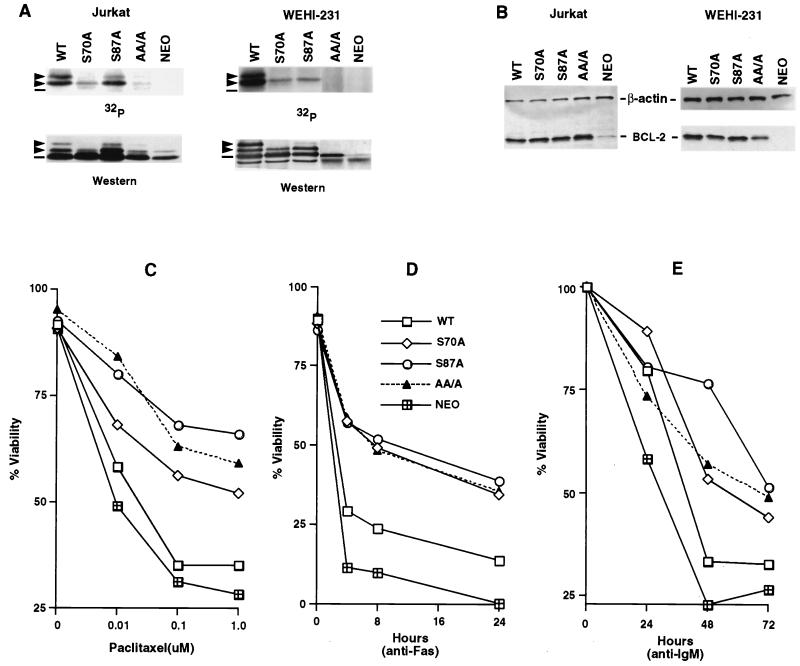
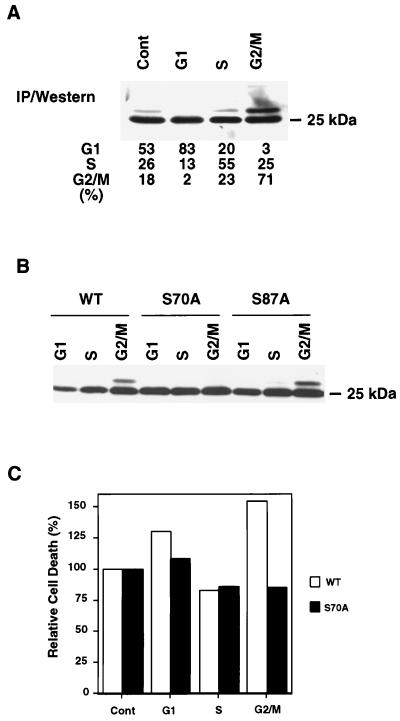
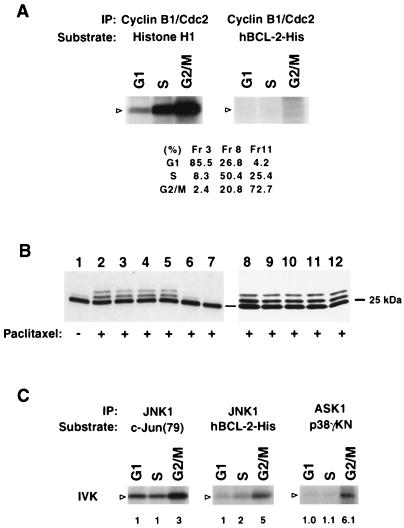
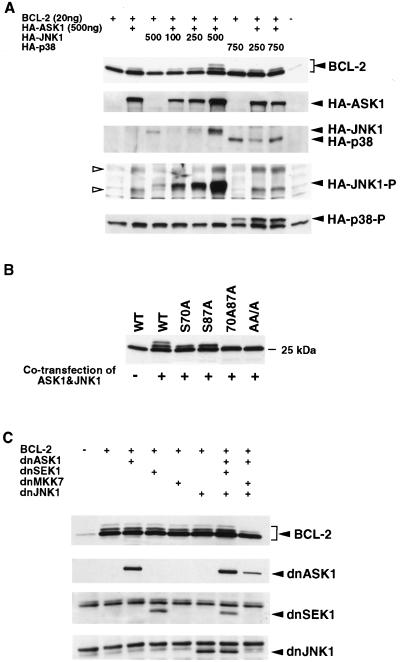
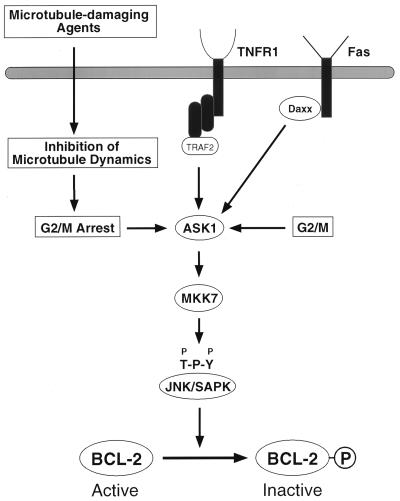
Multiple signal transduction pathways are capable of modifying BCL-2 family members to reset susceptibility to apoptosis. We used two-dimensional peptide mapping and sequencing to identify three residues (Ser70, Ser87, and Thr69) within the unstructured loop of BCL-2 that were phosphorylated in response to microtubule-damaging agents, which also arrest cells at G(2)/M. Changing these sites to alanine conferred more antiapoptotic activity on BCL-2 following physiologic death signals as well as paclitaxel, indicating that phosphorylation is inactivating. An examination of cycling cells enriched by elutriation for distinct phases of the cell cycle revealed that BCL-2 was phosphorylated at the G(2)/M phase of the cell cycle. G(2)/M-phase cells proved more susceptible to death signals, and phosphorylation of BCL-2 appeared to be responsible, as a Ser70Ala substitution restored resistance to apoptosis. We noted that ASK1 and JNK1 were normally activated at G(2)/M phase, and JNK was capable of phosphorylating BCL-2. Expression of a series of wild-type and dominant-negative kinases indicated an ASK1/Jun N-terminal protein kinase 1 (JNK1) pathway phosphorylated BCL-2 in vivo. Moreover, the combination of dominant negative ASK1, (dnASK1), dnMKK7, and dnJNK1 inhibited paclitaxel-induced BCL-2 phosphorylation. Thus, stress response kinases phosphorylate BCL-2 during cell cycle progression as a normal physiologic process to inactivate BCL-2 at G(2)/M.
Figures
Similar articles
-
A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation.Cancer Res. 2004 Mar 15;64(6):2134-42. doi: 10.1158/0008-5472.can-03-2296. Cancer Res. 2004. PMID: 15026354
-
Modulation of mitogen-activated protein kinases and phosphorylation of Bcl-2 by vinblastine represent persistent forms of normal fluctuations at G2-M1.Cancer Res. 2000 Nov 15;60(22):6403-7. Cancer Res. 2000. PMID: 11103805
-
Involvement of Asp-Glu-Val-Asp-directed, caspase-mediated mitogen-activated protein kinase kinase 1 Cleavage, c-Jun N-terminal kinase activation, and subsequent Bcl-2 phosphorylation for paclitaxel-induced apoptosis in HL-60 cells.Mol Pharmacol. 2001 Feb;59(2):254-62. doi: 10.1124/mol.59.2.254. Mol Pharmacol. 2001. PMID: 11160861
-
Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice.Antioxid Redox Signal. 2002 Jun;4(3):415-25. doi: 10.1089/15230860260196218. Antioxid Redox Signal. 2002. PMID: 12215209 Review.
-
PP2C family members play key roles in regulation of cell survival and apoptosis.Cancer Sci. 2006 Jul;97(7):563-7. doi: 10.1111/j.1349-7006.2006.00219.x. Cancer Sci. 2006. PMID: 16827794 Free PMC article. Review.
Cited by
-
Proteotoxic Stress and Cell Death in Cancer Cells.Cancers (Basel). 2020 Aug 23;12(9):2385. doi: 10.3390/cancers12092385. Cancers (Basel). 2020. PMID: 32842524 Free PMC article. Review.
-
The role of autophagy in metal-induced urogenital carcinogenesis.Semin Cancer Biol. 2021 Nov;76:247-257. doi: 10.1016/j.semcancer.2021.03.022. Epub 2021 Mar 30. Semin Cancer Biol. 2021. PMID: 33798723 Free PMC article. Review.
-
Early apoptosis in different models of cardiac hypertrophy induced by high renin-angiotensin system activity involves CaMKII.J Appl Physiol (1985). 2012 Jun;112(12):2110-20. doi: 10.1152/japplphysiol.01383.2011. Epub 2012 Apr 5. J Appl Physiol (1985). 2012. PMID: 22492934 Free PMC article.
-
Phospho-Bcl-x(L)(Ser62) plays a key role at DNA damage-induced G(2) checkpoint.Cell Cycle. 2012 Jun 1;11(11):2159-69. doi: 10.4161/cc.20672. Epub 2012 Jun 1. Cell Cycle. 2012. PMID: 22617334 Free PMC article.
-
Endoplasmic reticulum stress and type 2 diabetes.Annu Rev Biochem. 2012;81:767-93. doi: 10.1146/annurev-biochem-072909-095555. Epub 2012 Mar 23. Annu Rev Biochem. 2012. PMID: 22443930 Free PMC article. Review.
References
-
- Bakhshi A, Jensen J P, Goldman P, Wright J J, McBride O W, Epstein A L, Korsmeyer S J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. - PubMed
-
- Blagosklonny M V, Giannakakou P, el-Deiry W S, Kingston D G, Higgs P I, Neckers L, Fojo T. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res. 1997;57:130–135. - PubMed
-
- Blume-Jensen P, Janknecht R, Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr Biol. 1998;8:779–782. - PubMed
-
- Boyle W J, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous