

Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation
- PMID: 9566918
- PMCID: PMC110678
- DOI: 10.1128/MCB.18.5.2986
Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation
Abstract
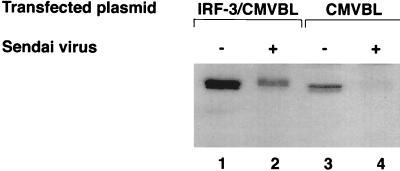
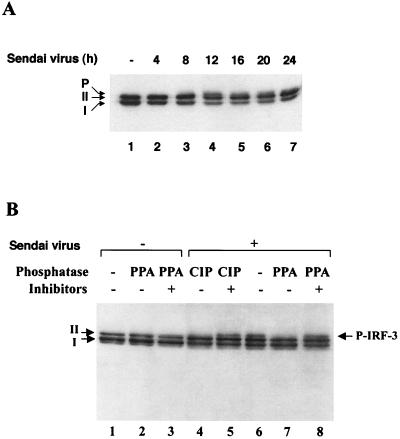
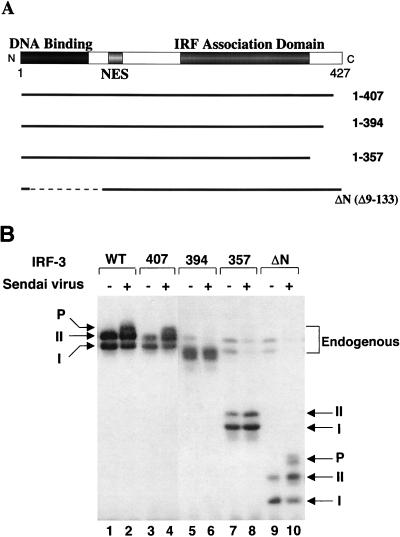
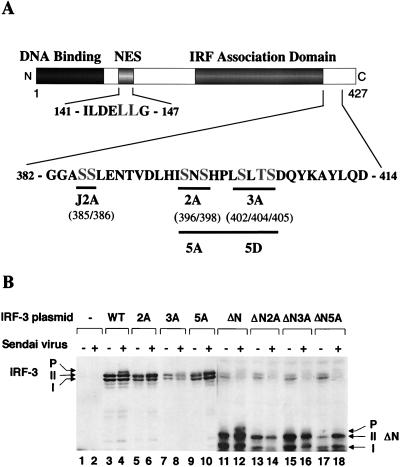
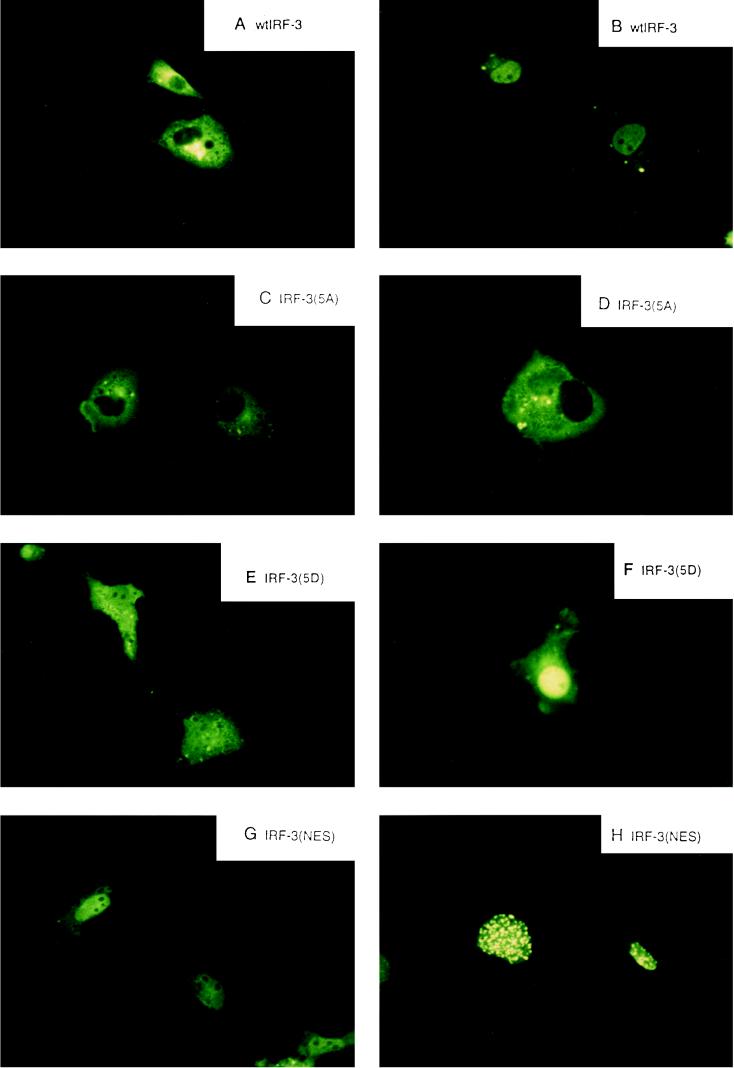
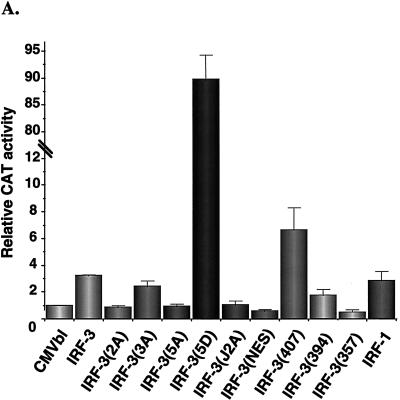
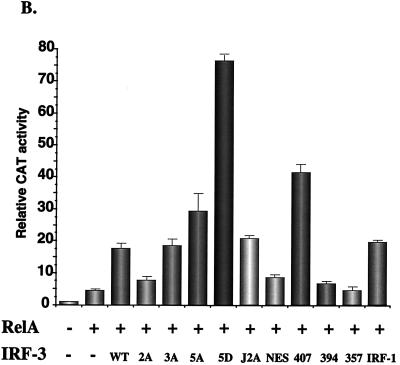
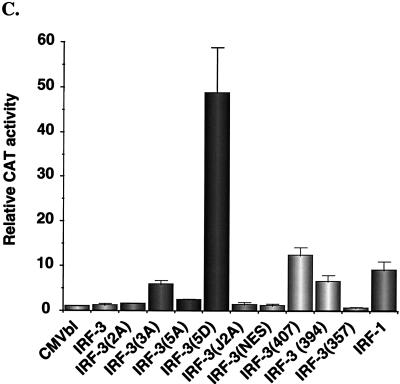
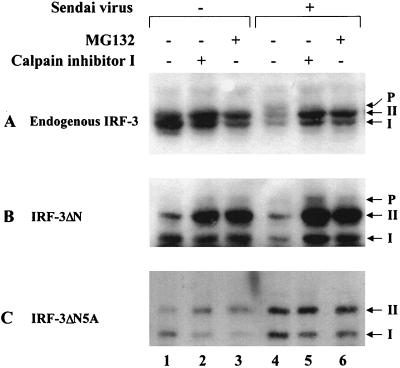
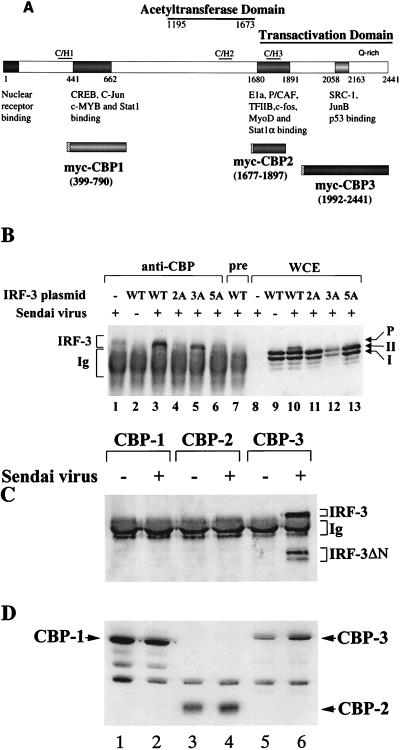
The interferon regulatory factors (IRF) consist of a growing family of related transcription proteins first identified as regulators of the alpha beta interferon (IFN-alpha/beta) gene promoters, as well as the interferon-stimulated response element (ISRE) of some IFN-stimulated genes. IRF-3 was originally identified as a member of the IRF family based on homology with other IRF family members and on binding to the ISRE of the ISG15 promoter. IRF-3 is expressed constitutively in a variety of tissues, and the relative levels of IRF-3 mRNA do not change in virus-infected or IFN-treated cells. In the present study, we demonstrate that following Sendai virus infection, IRF-3 is posttranslationally modified by protein phosphorylation at multiple serine and threonine residues, which are located in the carboxy terminus of IRF-3. A combination of IRF-3 deletion and point mutations localized the inducible phosphorylation sites to the region -ISNSHPLSLTSDQ- between amino acids 395 and 407; point mutation of residues Ser-396 and Ser-398 eliminated virus-induced phosphorylation of IRF-3 protein, although residues Ser-402, Thr-404, and Ser-405 were also targets. Phosphorylation results in the cytoplasm-to-nucleus translocation of IRF-3, DNA binding, and increased transcriptional activation. Substitution of the Ser-Thr sites with the phosphomimetic Asp generated a constitutively active form of IRF-3 that functioned as a very strong activator of promoters containing PRDI-PRDIII or ISRE regulatory elements. Phosphorylation also appears to represent a signal for virus-mediated degradation, since the virus-induced turnover of IRF-3 was prevented by mutation of the IRF-3 Ser-Thr cluster or by proteasome inhibitors. Interestingly, virus infection resulted in the association of IRF-3 with the CREB binding protein (CBP) coactivator, as detected by coimmunoprecipitation with anti-CBP antibody, an interaction mediated by the C-terminal domains of both proteins. Mutation of residues Ser-396 and Ser-398 in IRF-3 abrogated its binding to CBP. These results are discussed in terms of a model in which virus-inducible, C-terminal phosphorylation of IRF-3 alters protein conformation to permit nuclear translocation, association with transcriptional partners, and primary activation of IFN- and IFN-responsive genes.
Figures
Similar articles
-
Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains.Mol Cell Biol. 1999 Apr;19(4):2465-74. doi: 10.1128/MCB.19.4.2465. Mol Cell Biol. 1999. PMID: 10082512 Free PMC article.
-
Triggering the interferon response: the role of IRF-3 transcription factor.J Interferon Cytokine Res. 1999 Jan;19(1):1-13. doi: 10.1089/107999099314360. J Interferon Cytokine Res. 1999. PMID: 10048763 Review.
-
Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300.EMBO J. 1998 Feb 16;17(4):1087-95. doi: 10.1093/emboj/17.4.1087. EMBO J. 1998. PMID: 9463386 Free PMC article.
-
Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA.J Biol Chem. 2003 Mar 14;278(11):9441-7. doi: 10.1074/jbc.M209851200. Epub 2003 Jan 10. J Biol Chem. 2003. PMID: 12524442
-
On the role of IRF in host defense.J Interferon Cytokine Res. 2002 Jan;22(1):59-71. doi: 10.1089/107999002753452665. J Interferon Cytokine Res. 2002. PMID: 11846976 Review.
Cited by
-
DNA-PKcs restricts Zika virus spreading and is required for effective antiviral response.Front Immunol. 2022 Oct 13;13:1042463. doi: 10.3389/fimmu.2022.1042463. eCollection 2022. Front Immunol. 2022. PMID: 36311766 Free PMC article.
-
PLAAT1 inhibits type I interferon response via degradation of IRF3 and IRF7 in Zebrafish.Front Immunol. 2022 Sep 12;13:979919. doi: 10.3389/fimmu.2022.979919. eCollection 2022. Front Immunol. 2022. PMID: 36172355 Free PMC article.
-
African Swine Fever Virus Host-Pathogen Interactions.Subcell Biochem. 2023;106:283-331. doi: 10.1007/978-3-031-40086-5_11. Subcell Biochem. 2023. PMID: 38159232
-
Anti-viral and anti-inflammatory mechanisms of the innate immune transcription factor interferon regulatory factor 3: relevance to human CNS diseases.J Neuroimmune Pharmacol. 2013 Mar;8(1):132-44. doi: 10.1007/s11481-012-9360-5. Epub 2012 Jun 10. J Neuroimmune Pharmacol. 2013. PMID: 22684309 Review.
-
Poly (I:C), an agonist of toll-like receptor-3, inhibits replication of the Chikungunya virus in BEAS-2B cells.Virol J. 2012 Jun 14;9:114. doi: 10.1186/1743-422X-9-114. Virol J. 2012. PMID: 22698190 Free PMC article.
References
-
- Arany Z, Sellers W R, Livingston D M, Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77:799–800. - PubMed
-
- Avantaggiati M L, Ogryzko V, Gardner K, Giordano A, Levine A S, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. - PubMed
-
- Baldwin A S., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. - PubMed
-
- Bannister A J, Kouzarides T. The CBP coactivator is a histone acetyltransferase. Nature. 1996;384:641–643. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous