

Rapid, potent, and persistent covalent chemical probes to deconvolute PI3Kα signaling
- PMID: 39568927
- PMCID: PMC11575505
- DOI: 10.1039/d4sc05459h
Rapid, potent, and persistent covalent chemical probes to deconvolute PI3Kα signaling
Abstract
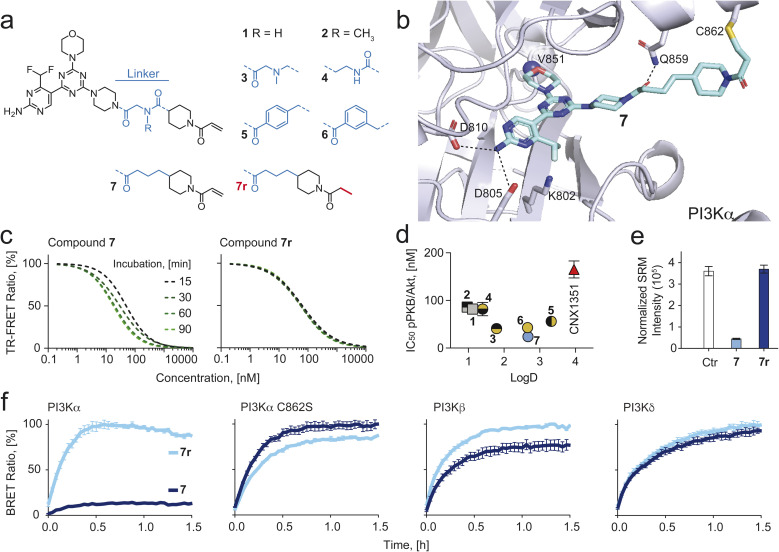
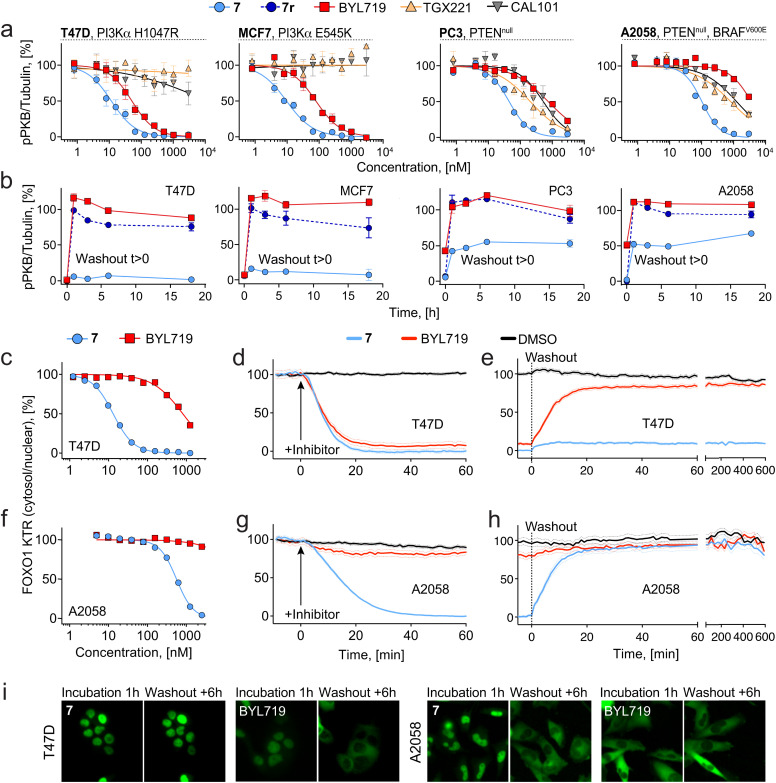
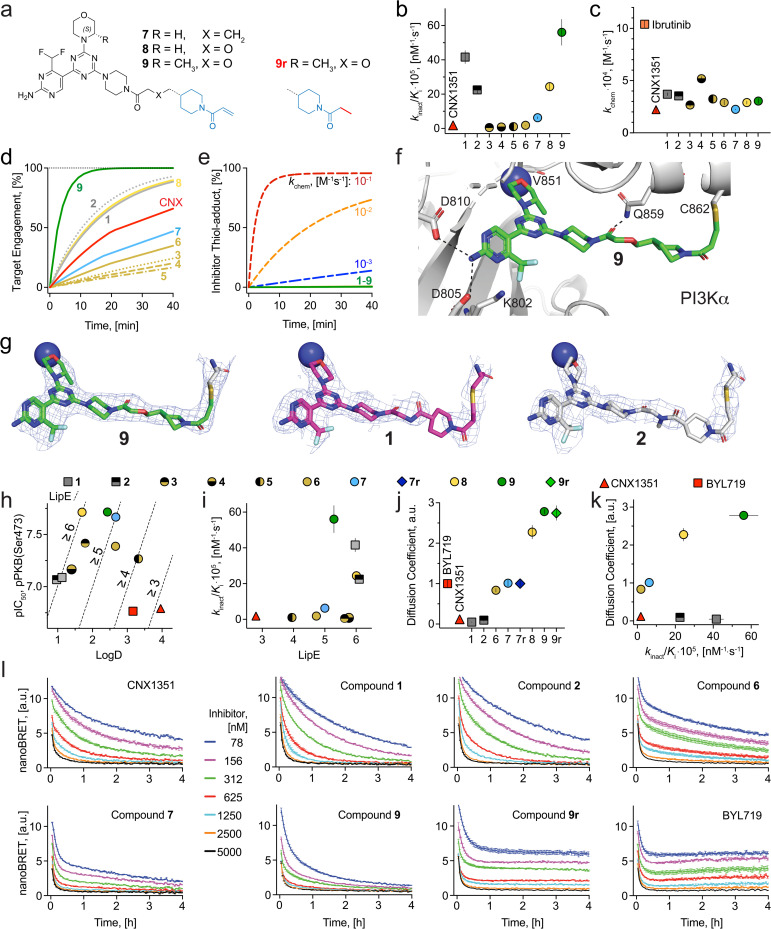
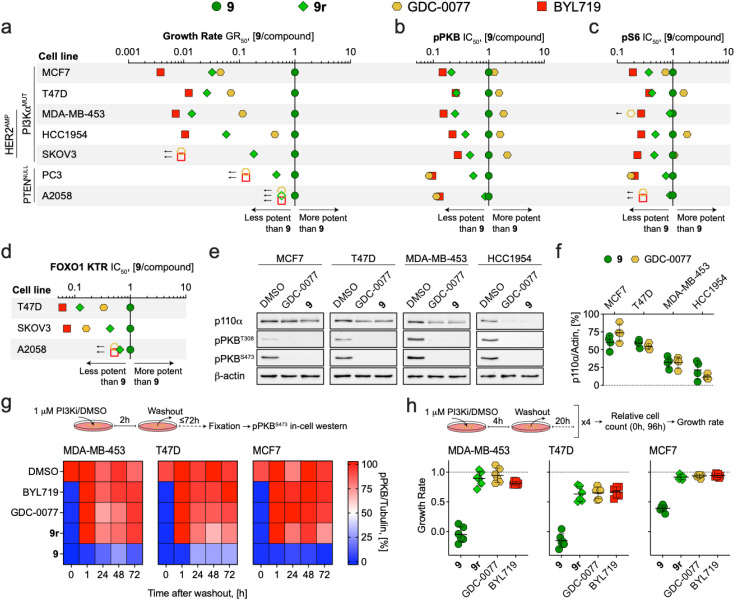
Chemical probes have gained importance in the elucidation of signal transduction in biology. Insufficient selectivity and potency, lack of cellular activity and inappropriate use of chemical probes has major consequences on interpretation of biological results. The catalytic subunit of phosphoinositide 3-kinase α (PI3Kα) is one of the most frequently mutated genes in cancer, but fast-acting, high-quality probes to define PI3Kα's specific function to clearly separate it from other class I PI3K isoforms, are not available. Here, we present a series of novel covalent PI3Kα-targeting probes with optimized intracellular target access and kinetic parameters. On-target TR-FRET and off-target assays provided relevant kinetic parameters (k chem, k inact and K i) to validate our chemical probes. Additional intracellular nanoBRET tracer displacement measurements showed rapid diffusion across the cell membrane and extremely fast target engagement, while investigations of signaling downstream of PI3Kα via protein kinase B (PKB/Akt) and forkhead box O (FOXO) revealed blunted pathway activity in cancer cell lines with constitutively activated PI3Kα lasting for several days. In contrast, persistent PI3Kα inhibition was rapidly bypassed by other class I PI3K isoforms in cells lacking functional phosphatase and tensin homolog (PTEN). Comparing the rapidly-diffusing, fast target-engaging chemical probe 9 to clinical reversible PI3Kα-selective inhibitors alpelisib, inavolisib and 9r, a reversible analogue of 9, revealed 9's superior potency to inhibit growth (up to 600-fold) associated with sustained suppression of PI3Kα signaling in breast cancer cell lines. Finally, using a simple washout protocol, the utility of the highly-selective covalent PI3Kα probe 9 was demonstrated by the quantification of the coupling of insulin, EGF and CXCL12 receptors to distinct PI3K isoforms for signal transduction in response to ligand-dependent activation. Collectively, these findings along with the novel covalent chemical probes against PI3Kα provide insights into isoform-specific functions in cancer cells and highlight opportunities to achieve improved selectivity and long-lasting efficacy.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
MPW is founder and shareholder of Akylox Therapeutics ApS.
Figures

Similar articles
-
Enabling Systemic Identification and Functionality Profiling for Cdc42 Homeostatic Modulators.bioRxiv [Preprint]. 2024 Jan 8:2024.01.05.574351. doi: 10.1101/2024.01.05.574351. bioRxiv. 2024. Update in: Commun Chem. 2024 Nov 19;7(1):271. doi: 10.1038/s42004-024-01352-7. PMID: 38260445 Free PMC article. Updated. Preprint.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Falls prevention interventions for community-dwelling older adults: systematic review and meta-analysis of benefits, harms, and patient values and preferences.Syst Rev. 2024 Nov 26;13(1):289. doi: 10.1186/s13643-024-02681-3. Syst Rev. 2024. PMID: 39593159 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Impact of residual disease as a prognostic factor for survival in women with advanced epithelial ovarian cancer after primary surgery.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD015048. doi: 10.1002/14651858.CD015048.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161421 Free PMC article. Review.
References
-
- Blagg J. Workman P. Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology. Cancer Cell. 2017;32:9–25. doi: 10.1016/j.ccell.2017.06.005. https://dx.doi.org/10.1016/j.ccell.2017.06.005 - DOI - DOI - PMC - PubMed
-
- Vu V. Szewczyk M. M. Nie D. Y. Arrowsmith C. H. Barsyte-Lovejoy D. Validating Small Molecule Chemical Probes for Biological Discovery. Annu. Rev. Biochem. 2022;91:61–87. doi: 10.1146/annurev-biochem-032620-105344. https://dx.doi.org/10.1146/annurev-biochem-032620-105344 - DOI - DOI - PubMed
-
- Garbaccio R. M. Parmee E. R. The Impact of Chemical Probes in Drug Discovery: A Pharmaceutical Industry Perspective. Cell Chem. Biol. 2016;23:10–17. doi: 10.1016/j.chembiol.2015.11.011. https://dx.doi.org/10.1016/j.chembiol.2015.11.011 - DOI - DOI - PubMed
-
- Bunnage M. E. Chekler E. L. Jones L. H. Target validation using chemical probes. Nat. Chem. Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. https://dx.doi.org/10.1038/nchembio.1197 - DOI - DOI - PubMed
-
- Arrowsmith C. H. Audia J. E. Austin C. Baell J. Bennett J. Blagg J. Bountra C. Brennan P. E. Brown P. J. Bunnage M. E. Buser-Doepner C. Campbell R. M. Carter A. J. Cohen P. Copeland R. A. Cravatt B. Dahlin J. L. Dhanak D. Edwards A. M. Frederiksen M. Frye S. V. Gray N. Grimshaw C. E. Hepworth D. Howe T. Huber K. V. Jin J. Knapp S. Kotz J. D. Kruger R. G. Lowe D. Mader M. M. Marsden B. Mueller-Fahrnow A. Müller S. O'Hagan R. C. Overington J. P. Owen D. R. Rosenberg S. H. Roth B. Roth B. Ross R. Schapira M. Schreiber S. L. Shoichet B. Sundström M. Superti-Furga G. Taunton J. Toledo-Sherman L. Walpole C. Walters M. A. Willson T. M. Workman P. Young R. N. Zuercher W. J. The promise and peril of chemical probes. Nat. Chem. Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. https://dx.doi.org/10.1038/nchembio.1867 - DOI - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous