

Genomic instability as a driver and suppressor of anti-tumor immunity
- PMID: 39544936
- PMCID: PMC11562473
- DOI: 10.3389/fimmu.2024.1462496
Genomic instability as a driver and suppressor of anti-tumor immunity
Abstract
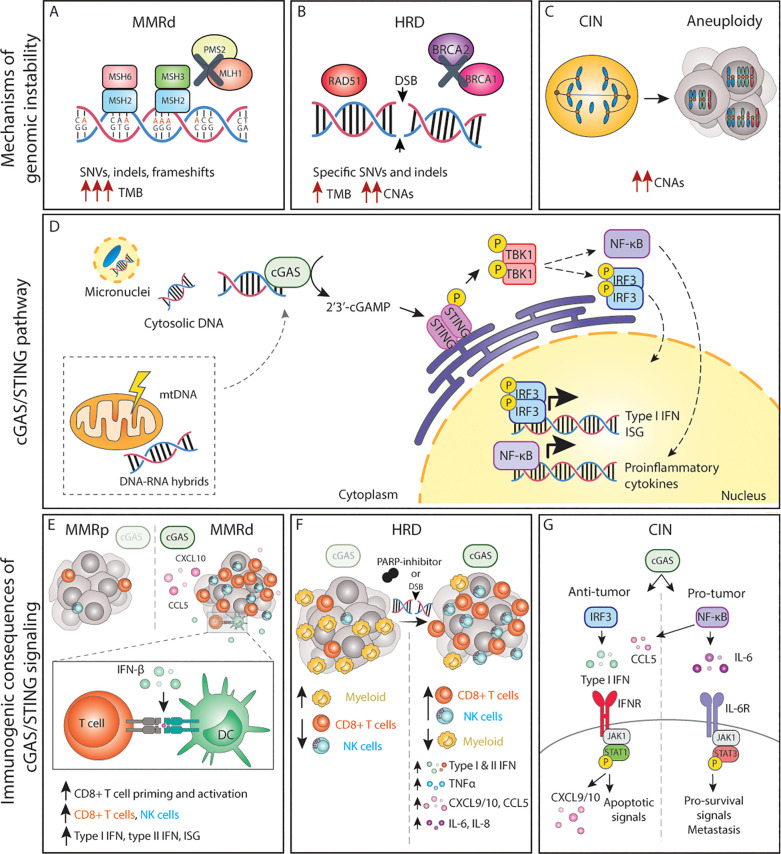
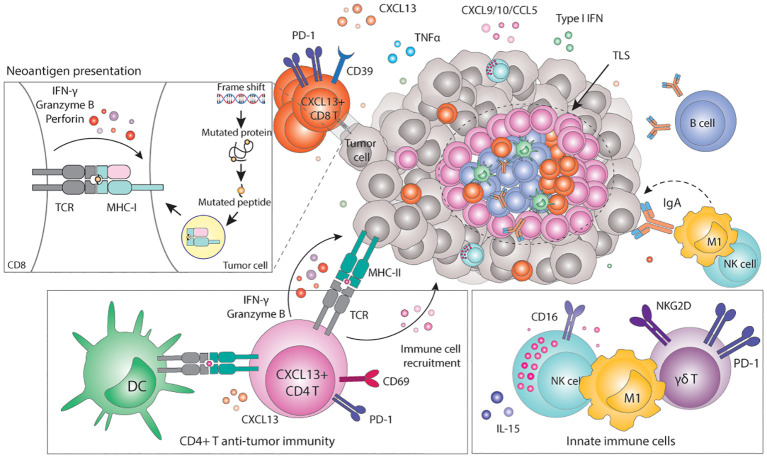
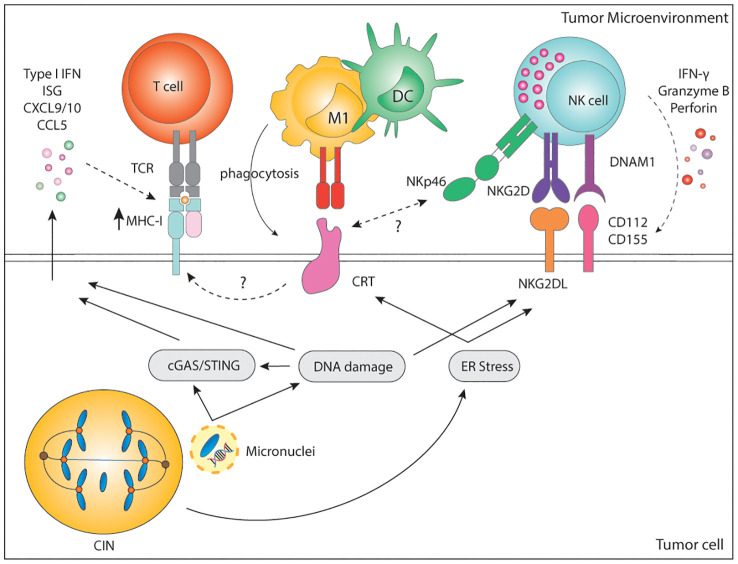
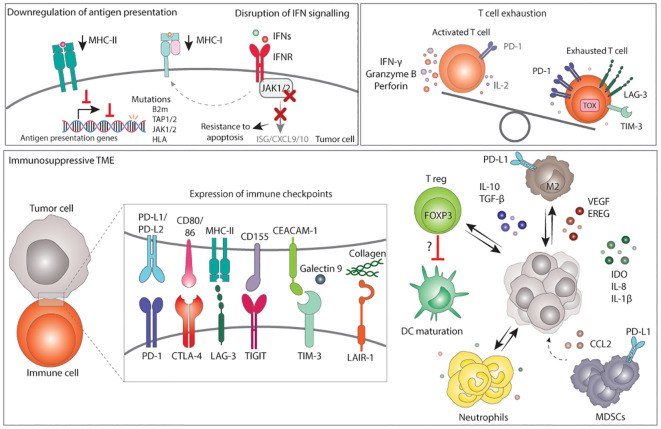
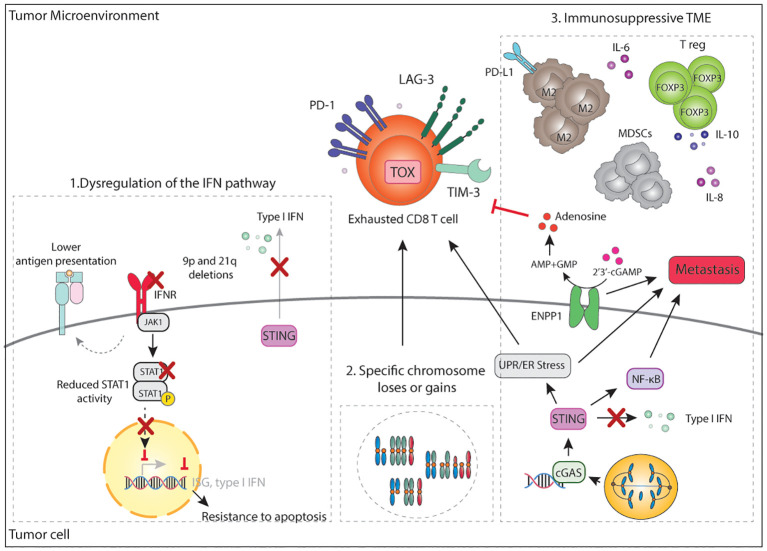
Genomic instability is a driver and accelerator of tumorigenesis and influences disease outcomes across cancer types. Although genomic instability has been associated with immune evasion and worsened disease prognosis, emerging evidence shows that genomic instability instigates pro-inflammatory signaling and enhances the immunogenicity of tumor cells, making them more susceptible to immune recognition. While this paradoxical role of genomic instability in cancer is complex and likely context-dependent, understanding it is essential for improving the success rates of cancer immunotherapy. In this review, we provide an overview of the underlying mechanisms that link genomic instability to pro-inflammatory signaling and increased immune surveillance in the context of cancer, as well as discuss how genomically unstable tumors evade the immune system. A better understanding of the molecular crosstalk between genomic instability, inflammatory signaling, and immune surveillance could guide the exploitation of immunotherapeutic vulnerabilities in cancer.
Keywords: MMRd; cGAS-STING; chromosomal instability; genomic instability; immune evasion; tumor-infiltrating lymphocytes.
Copyright © 2024 Requesens, Foijer, Nijman and de Bruyn.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Inflammatory signaling in genomically instable cancers.Cell Cycle. 2019 Aug;18(16):1830-1848. doi: 10.1080/15384101.2019.1638192. Epub 2019 Jul 10. Cell Cycle. 2019. PMID: 31260383 Free PMC article. Review.
-
cGAS-STING signalling in cancer: striking a balance with chromosomal instability.Biochem Soc Trans. 2023 Apr 26;51(2):539-555. doi: 10.1042/BST20220838. Biochem Soc Trans. 2023. PMID: 36876871 Free PMC article. Review.
-
Bidirectional regulation of the cGAS-STING pathway in the immunosuppressive tumor microenvironment and its association with immunotherapy.Front Immunol. 2024 Oct 11;15:1470468. doi: 10.3389/fimmu.2024.1470468. eCollection 2024. Front Immunol. 2024. PMID: 39464890 Free PMC article. Review.
-
The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability.Cells. 2019 Oct 10;8(10):1228. doi: 10.3390/cells8101228. Cells. 2019. PMID: 31658669 Free PMC article. Review.
-
Genomic instability, inflammatory signaling and response to cancer immunotherapy.Biochim Biophys Acta Rev Cancer. 2022 Jan;1877(1):188661. doi: 10.1016/j.bbcan.2021.188661. Epub 2021 Nov 17. Biochim Biophys Acta Rev Cancer. 2022. PMID: 34800547 Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials