

A stepwise guide for pangenome development in crop plants: an alfalfa (Medicago sativa) case study
- PMID: 39482604
- PMCID: PMC11526573
- DOI: 10.1186/s12864-024-10931-w
A stepwise guide for pangenome development in crop plants: an alfalfa (Medicago sativa) case study
Abstract
Background: The concept of pangenomics and the importance of structural variants is gaining recognition within the plant genomics community. Due to advancements in sequencing and computational technology, it has become feasible to sequence the entire genome of numerous individuals of a single species at a reasonable cost. Pangenomes have been constructed for many major diploid crops, including rice, maize, soybean, sorghum, pearl millet, peas, sunflower, grapes, and mustards. However, pangenomes for polyploid species are relatively scarce and are available in only few crops including wheat, cotton, rapeseed, and potatoes.
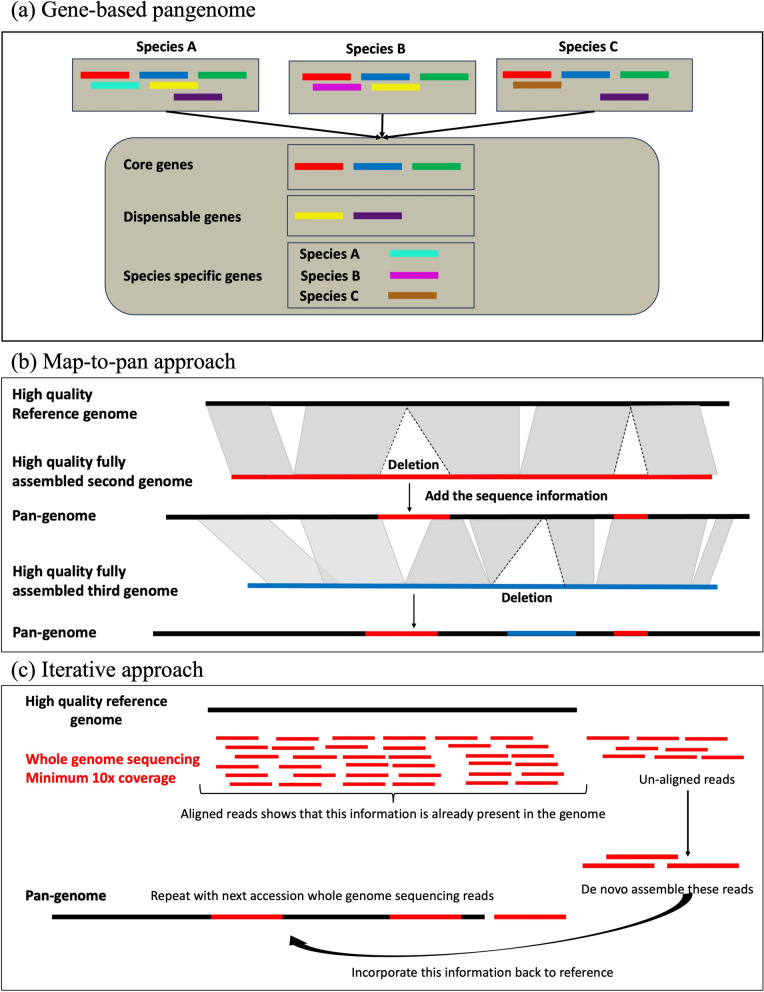
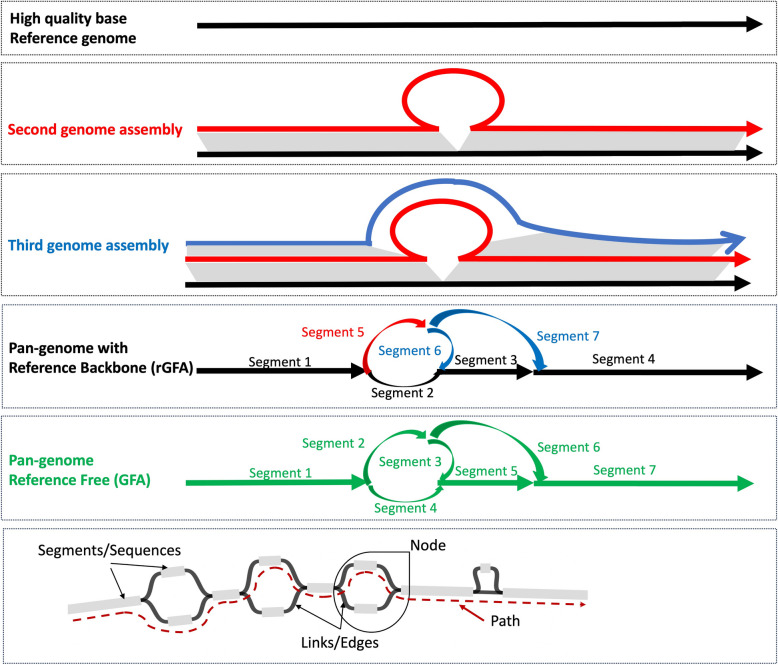
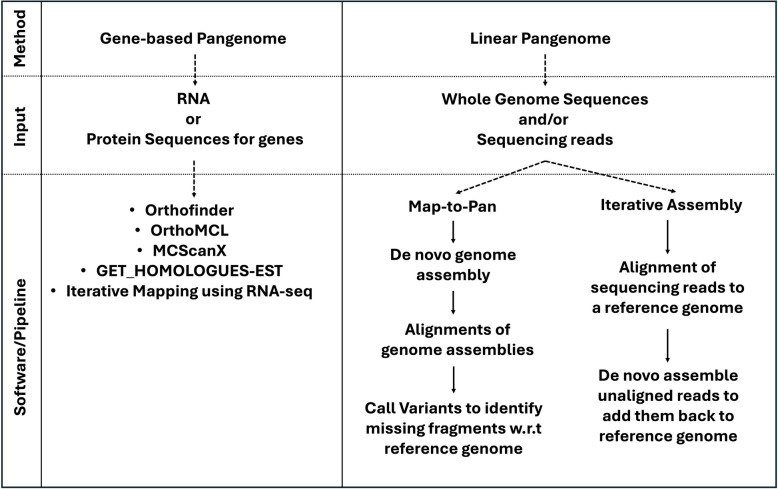
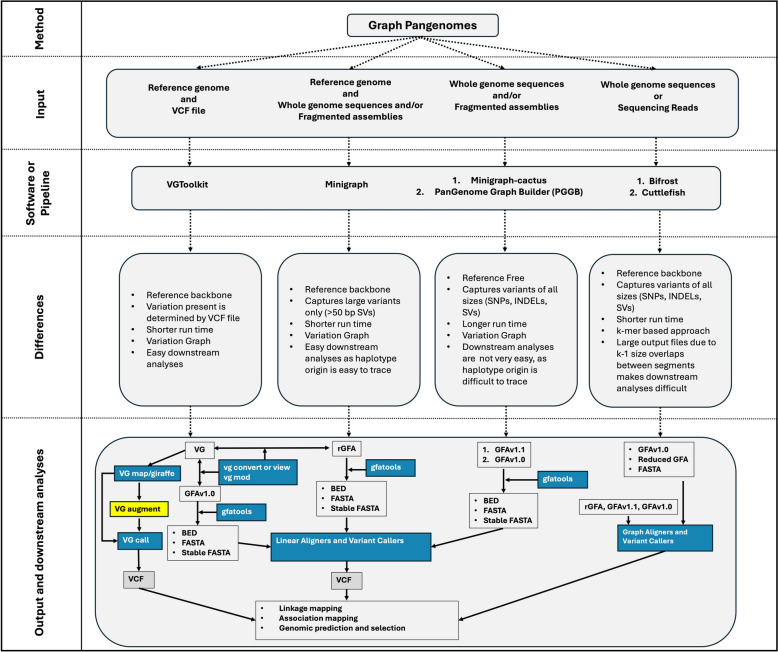
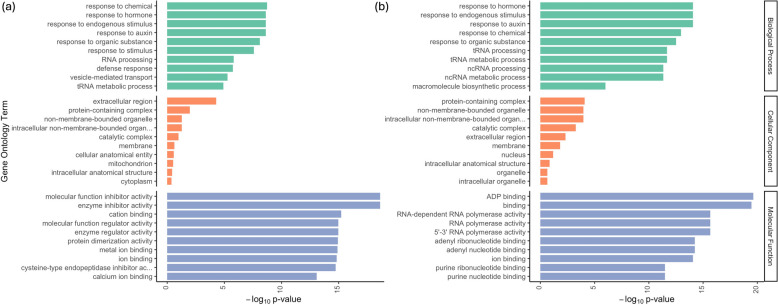
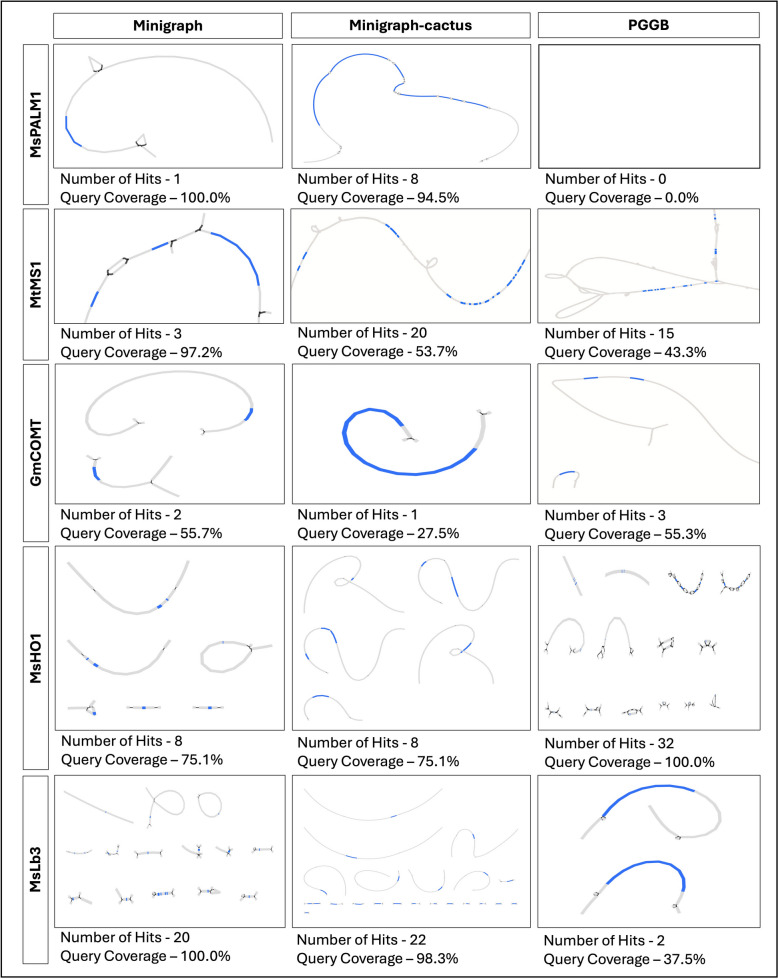
Main body: In this review, we explore the various methods used in crop pangenome development, discussing the challenges and implications of these techniques based on insights from published pangenome studies. We offer a systematic guide and discuss the tools available for constructing a pangenome and conducting downstream analyses. Alfalfa, a highly heterozygous, cross pollinated and autotetraploid forage crop species, is used as an example to discuss the concerns and challenges offered by polyploid crop species. We conducted a comparative analysis using linear and graph-based methods by constructing an alfalfa graph pangenome using three publicly available genome assemblies. To illustrate the intricacies captured by pangenome graphs for a complex crop genome, we used five different gene sequences and aligned them against the three graph-based pangenomes. The comparison of the three graph pangenome methods reveals notable variations in the genomic variation captured by each pipeline.
Conclusion: Pangenome resources are proving invaluable by offering insights into core and dispensable genes, novel gene discovery, and genome-wide patterns of variation. Developing user-friendly online portals for linear pangenome visualization has made these resources accessible to the broader scientific and breeding community. However, challenges remain with graph-based pangenomes including compatibility with other tools, extraction of sequence for regions of interest, and visualization of genetic variation captured in pangenome graphs. These issues necessitate further refinement of tools and pipelines to effectively address the complexities of polyploid, highly heterozygous, and cross-pollinated species.
Keywords: Alfalfa; Autotetraploid; Crop pangenome; Graph-based pangenome; Polyploids.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Pangenomes as a Resource to Accelerate Breeding of Under-Utilised Crop Species.Int J Mol Sci. 2022 Feb 28;23(5):2671. doi: 10.3390/ijms23052671. Int J Mol Sci. 2022. PMID: 35269811 Free PMC article. Review.
-
Pangenomics in crop improvement-from coding structural variations to finding regulatory variants with pangenome graphs.Plant Genome. 2022 Mar;15(1):e20177. doi: 10.1002/tpg2.20177. Epub 2021 Dec 13. Plant Genome. 2022. PMID: 34904403 Review.
-
A pangenome analysis pipeline provides insights into functional gene identification in rice.Genome Biol. 2023 Jan 26;24(1):19. doi: 10.1186/s13059-023-02861-9. Genome Biol. 2023. PMID: 36703158 Free PMC article.
-
Pangenome graphs in infectious disease: a comprehensive genetic variation analysis of Neisseria meningitidis leveraging Oxford Nanopore long reads.Front Genet. 2023 Aug 10;14:1225248. doi: 10.3389/fgene.2023.1225248. eCollection 2023. Front Genet. 2023. PMID: 37636268 Free PMC article.
-
Crop pangenomes.Vavilovskii Zhurnal Genet Selektsii. 2021 Feb;25(1):57-63. doi: 10.18699/VJ21.007. Vavilovskii Zhurnal Genet Selektsii. 2021. PMID: 34901703 Free PMC article.
References
-
- Sun S, Zhou Y, Chen J, Shi J, Zhao H, Zhao H, et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat Genet. 2018;50:1289–95. - PubMed
-
- Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet. 2019;51:1052–9. - PubMed
-
- Ge F, Qu J, Liu P, Pan L, Zou C, Yuan G, et al. Genome assembly of the maize inbred line A188 provides a new reference genome for functional genomics. Crop J. 2022;10:47–55.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
