

Multiscale molecular modeling of chromatin with MultiMM: From nucleosomes to the whole genome
- PMID: 39435339
- PMCID: PMC11492436
- DOI: 10.1016/j.csbj.2024.09.025
Multiscale molecular modeling of chromatin with MultiMM: From nucleosomes to the whole genome
Abstract
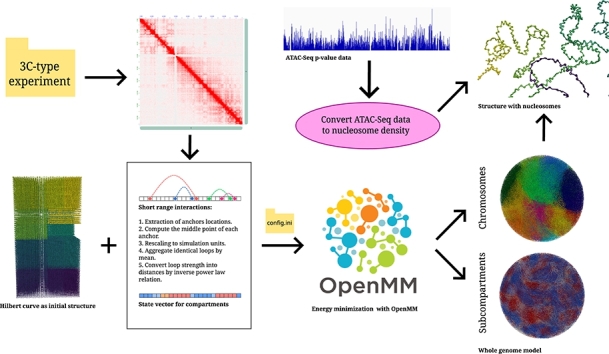
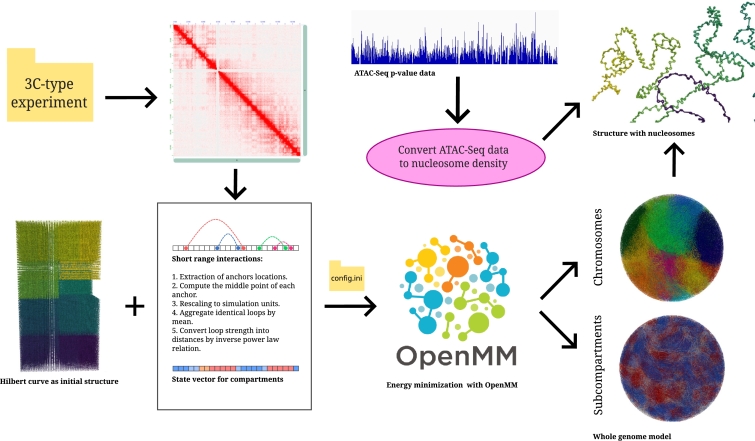
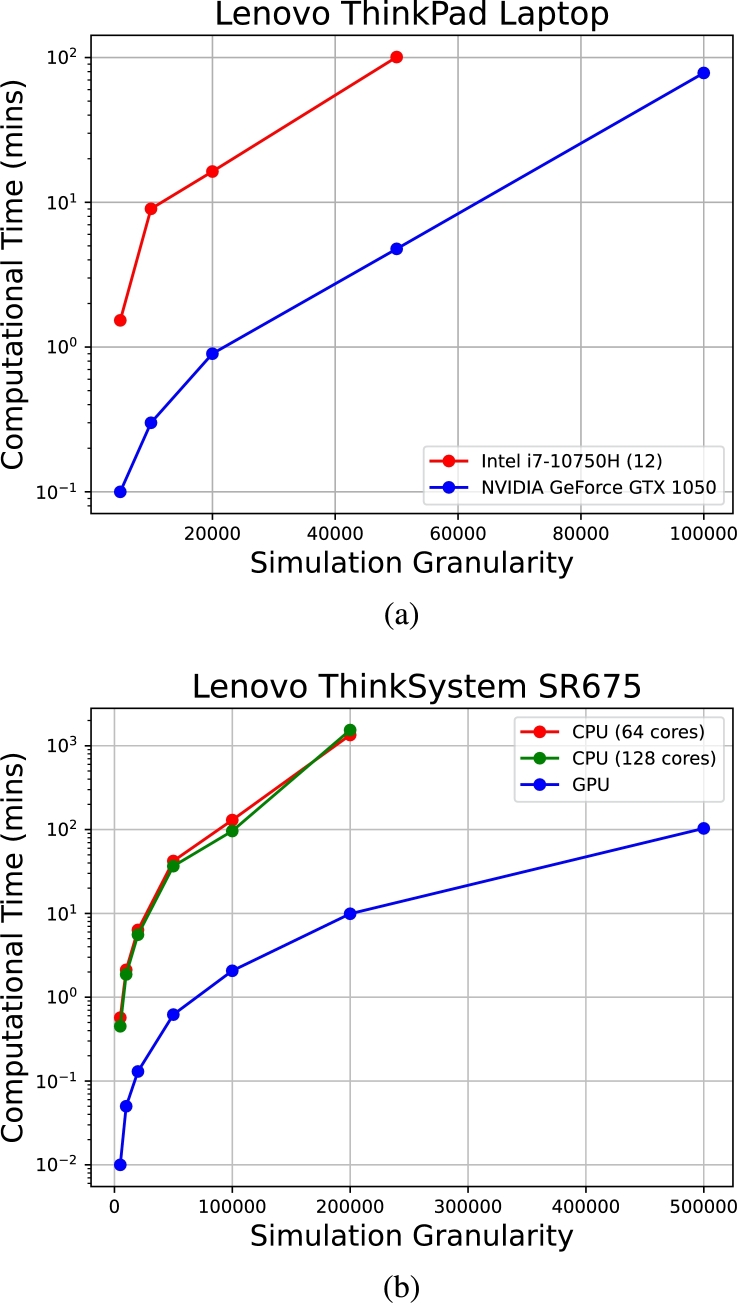
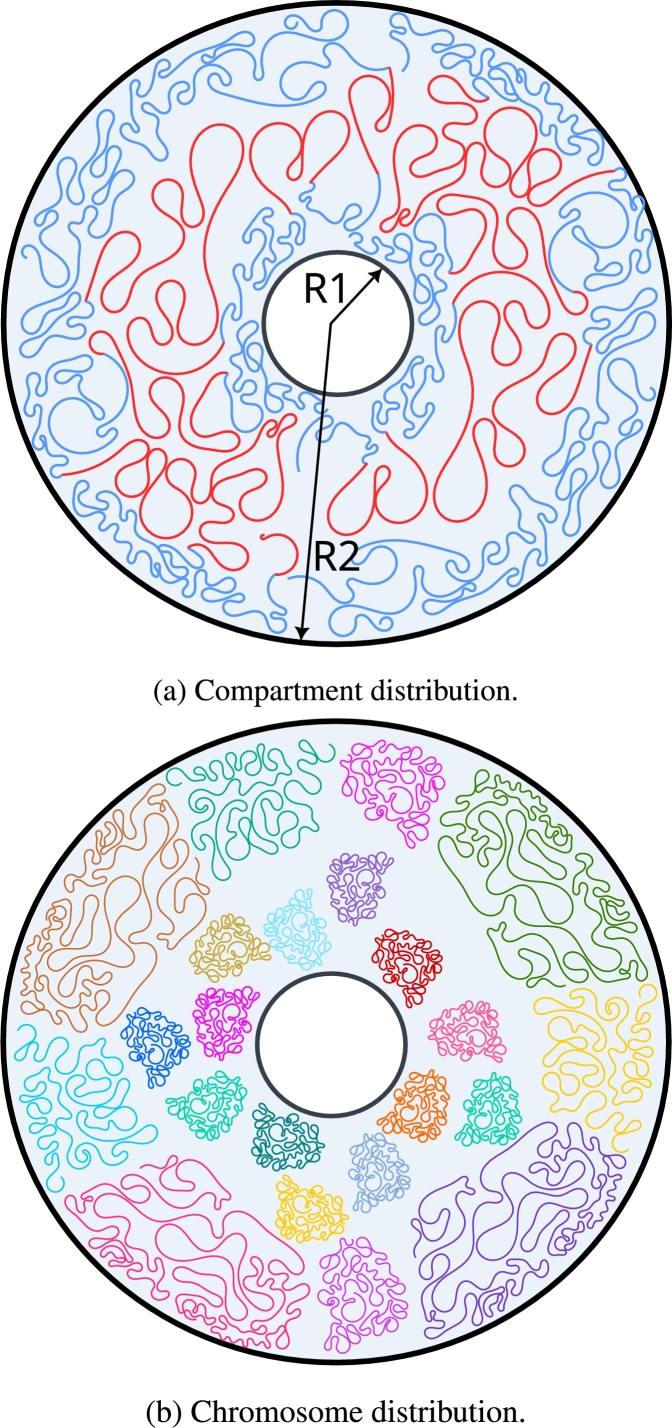
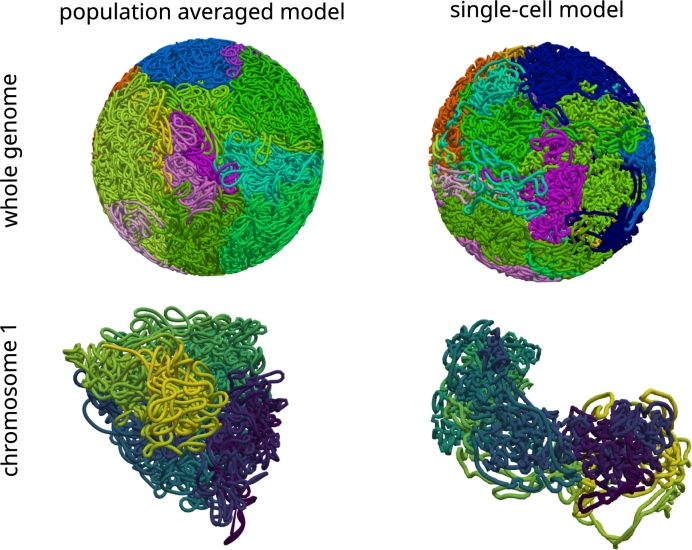
Motivation: We present a user-friendly 3D chromatin simulation model for the human genome based on OpenMM, addressing the challenges posed by existing models with use-specific implementations. Our approach employs a multi-scale energy minimization strategy, capturing chromatin's hierarchical structure. Initiating with a Hilbert curve-based structure, users can input files specifying nucleosome positioning, loops, compartments, or subcompartments. Results: The model utilizes an energy minimization approach with a large choice of numerical integrators, providing the entire genome's structure within minutes. Output files include the generated structures for each chromosome, offering a versatile and accessible tool for chromatin simulation in bioinformatics studies. Furthermore, MultiMM is capable of producing nucleosome-resolution structures by making simplistic geometric assumptions about the structure and the density of nucleosomes on the DNA. Code availability: Open-source software and the manual are freely available on https://github.com/SFGLab/MultiMM or via pip https://pypi.org/project/MultiMM/.
© 2024 The Authors.
Conflict of interest statement
Authors do not acknowledge any conflicts of interest.
Figures
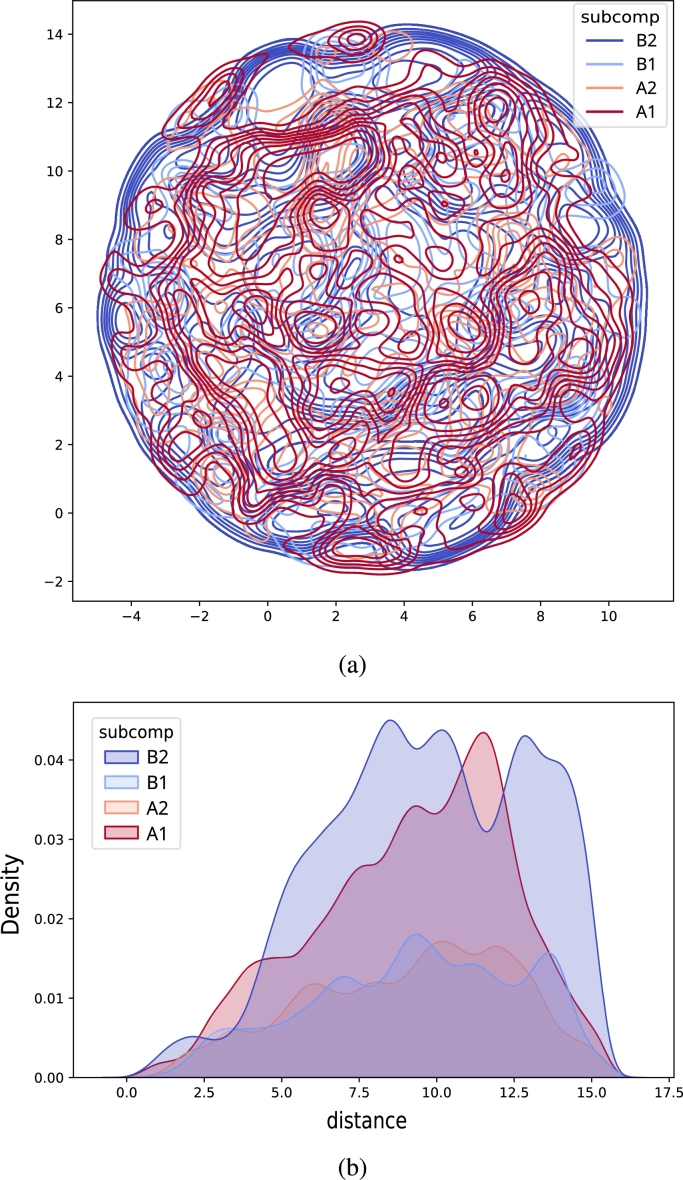
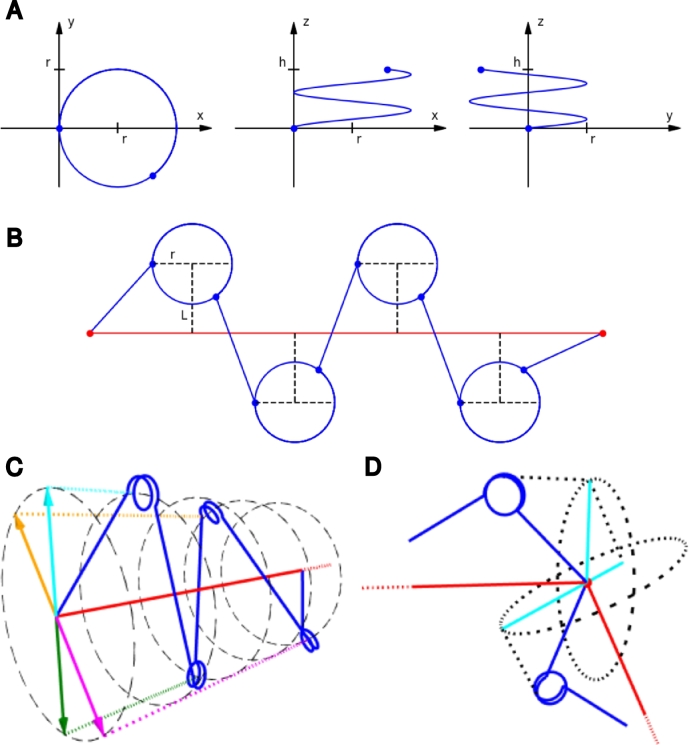
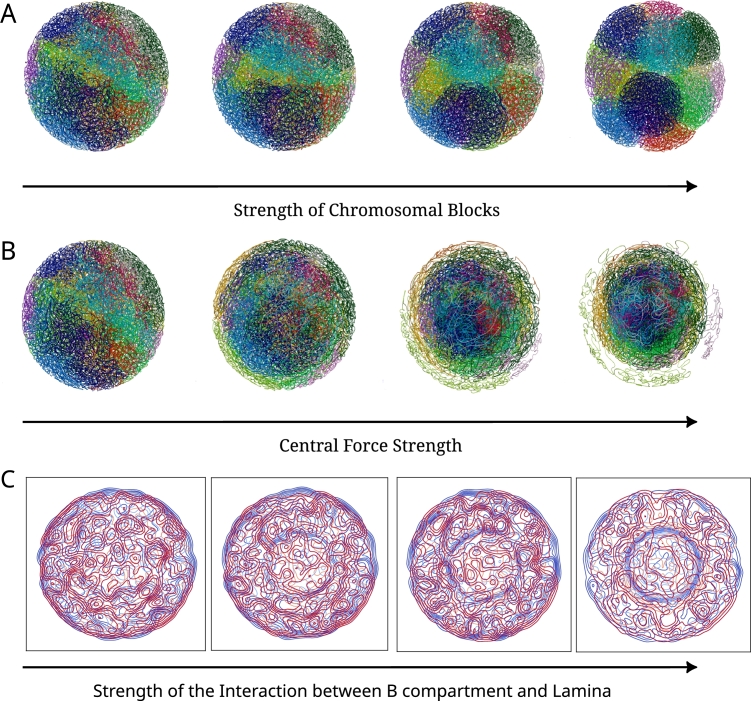
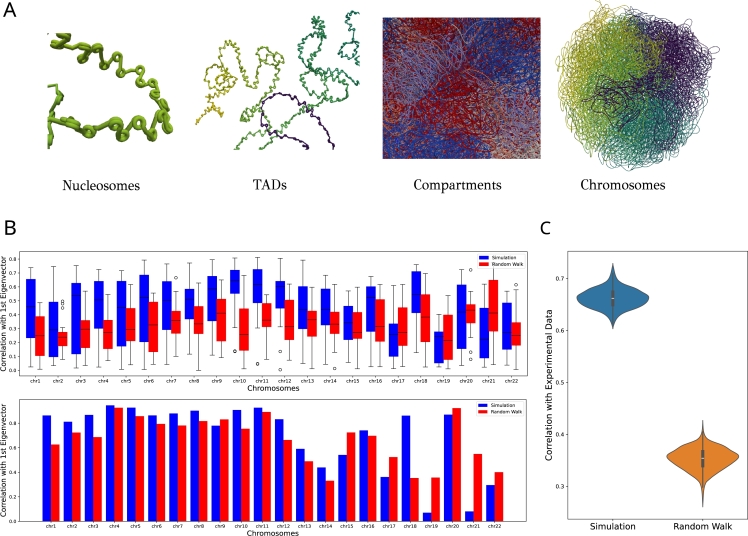
Similar articles
-
cudaMMC: GPU-enhanced multiscale Monte Carlo chromatin 3D modelling.Bioinformatics. 2023 Oct 3;39(10):btad588. doi: 10.1093/bioinformatics/btad588. Bioinformatics. 2023. PMID: 37774005 Free PMC article.
-
Predicting nucleosome positioning using a duration Hidden Markov Model.BMC Bioinformatics. 2010 Jun 24;11:346. doi: 10.1186/1471-2105-11-346. BMC Bioinformatics. 2010. PMID: 20576140 Free PMC article.
-
iNucs: inter-nucleosome interactions.Bioinformatics. 2021 Dec 7;37(23):4562-4563. doi: 10.1093/bioinformatics/btab698. Bioinformatics. 2021. PMID: 34623394 Free PMC article.
-
Bottom-Up Meets Top-Down: The Crossroads of Multiscale Chromatin Modeling.Biophys J. 2020 May 5;118(9):2057-2065. doi: 10.1016/j.bpj.2020.03.014. Epub 2020 Apr 4. Biophys J. 2020. PMID: 32320675 Free PMC article. Review.
-
[Attraction of Likenesses: Mechanisms of Self-Association and Compartmentalization of Eukaryotic Chromatin].Mol Biol (Mosk). 2019 Nov-Dec;53(6):933-953. doi: 10.1134/S0026898419060053. Mol Biol (Mosk). 2019. PMID: 31876274 Review. Russian.
References
-
- Bianco S., Chiariello A.M., Annunziatella C., Esposito A., Nicodemi M. Predicting chromatin architecture from models of polymer physics. Chromosom Res. 2017;25:25–34. - PubMed
-
- Biswas M., Langowski J., Bishop T.C. Atomistic simulations of nucleosomes. Wiley Interdiscip Rev Comput Mol Sci. 2013;3(4):378–392.
LinkOut - more resources
Full Text Sources