

Graphasing: phasing diploid genome assembly graphs with single-cell strand sequencing
- PMID: 39390579
- PMCID: PMC11466045
- DOI: 10.1186/s13059-024-03409-1
Graphasing: phasing diploid genome assembly graphs with single-cell strand sequencing
Abstract
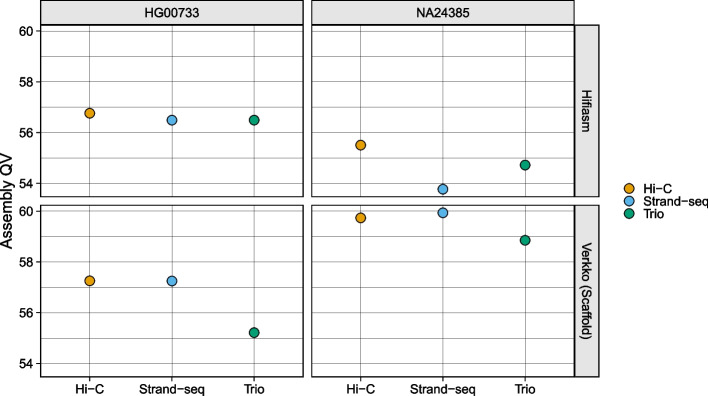
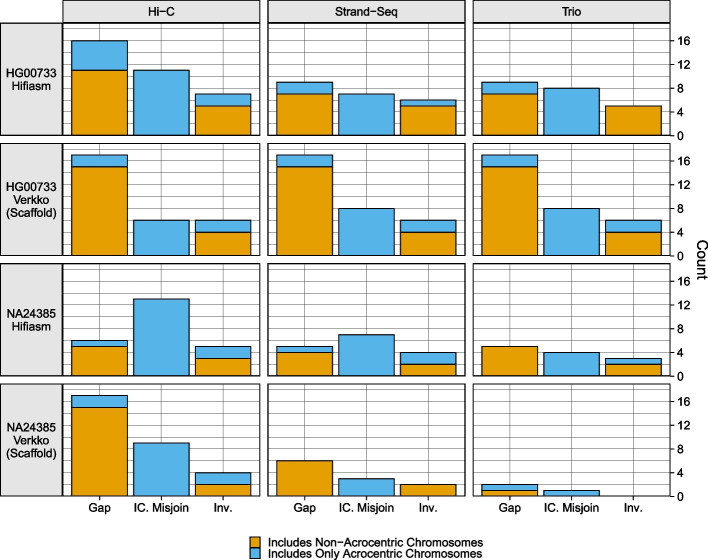
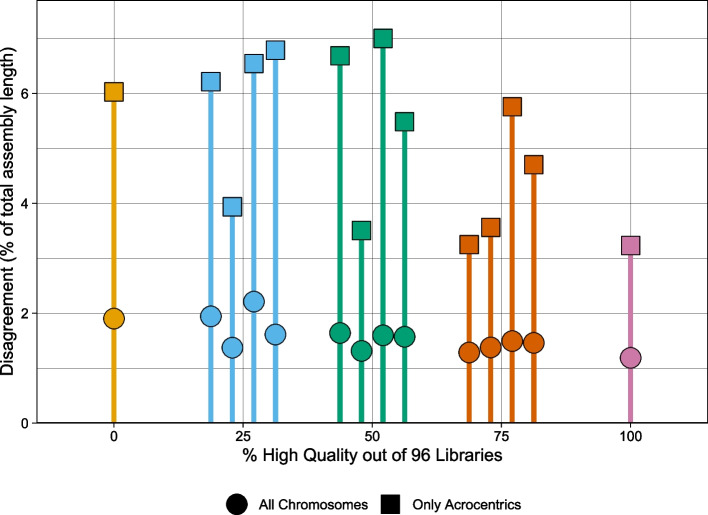
Haplotype information is crucial for biomedical and population genetics research. However, current strategies to produce de novo haplotype-resolved assemblies often require either difficult-to-acquire parental data or an intermediate haplotype-collapsed assembly. Here, we present Graphasing, a workflow which synthesizes the global phase signal of Strand-seq with assembly graph topology to produce chromosome-scale de novo haplotypes for diploid genomes. Graphasing readily integrates with any assembly workflow that both outputs an assembly graph and has a haplotype assembly mode. Graphasing performs comparably to trio phasing in contiguity, phasing accuracy, and assembly quality, outperforms Hi-C in phasing accuracy, and generates human assemblies with over 18 chromosome-spanning haplotypes.
Keywords: Assembly graph; De novo assembly; Haplotype; Hi-C; Hifiasm; Phasing; Strand-seq; Trio; Verkko.
© 2024. The Author(s).
Conflict of interest statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. S.K. has received travel funding for speaking at events hosted by ONT.
Figures
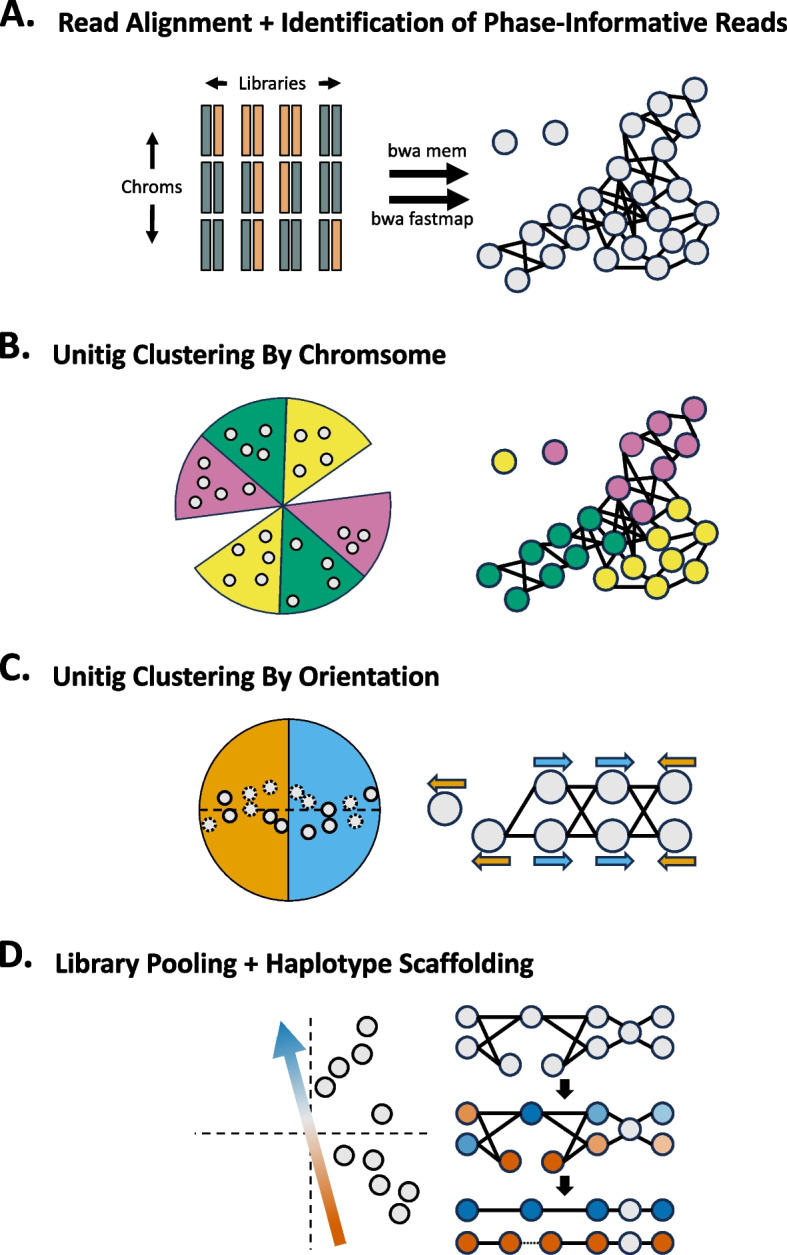
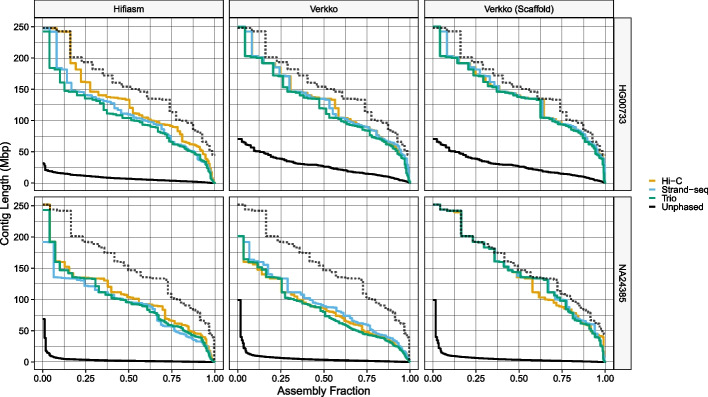
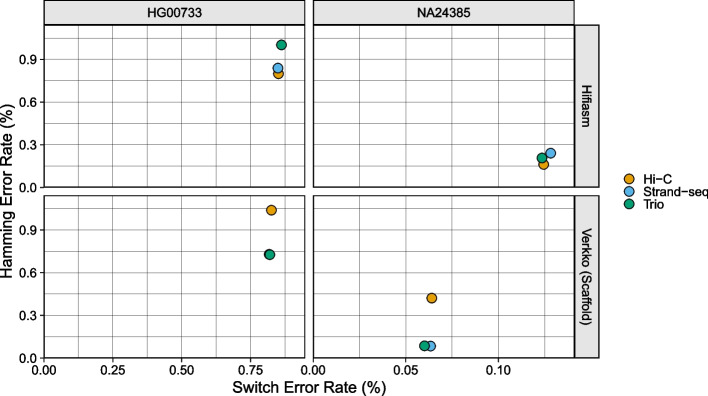
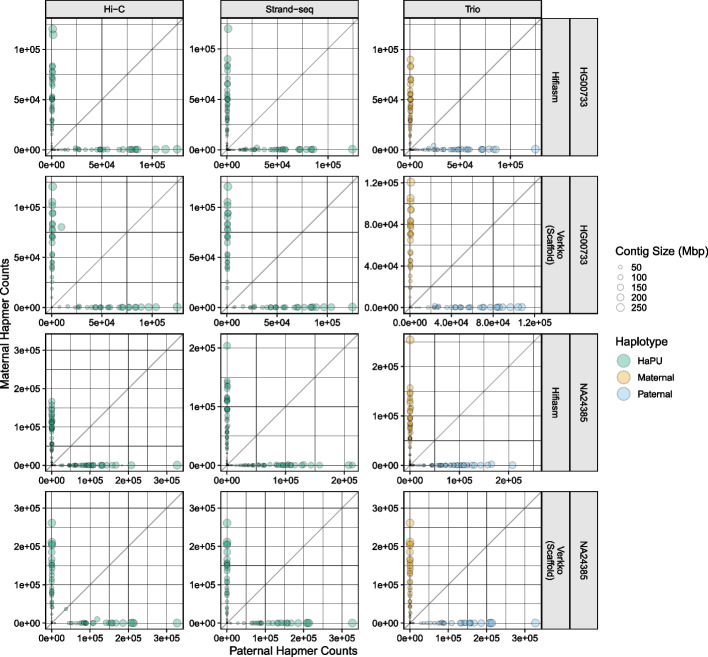

Update of
-
Phasing Diploid Genome Assembly Graphs with Single-Cell Strand Sequencing.bioRxiv [Preprint]. 2024 Jun 20:2024.02.15.580432. doi: 10.1101/2024.02.15.580432. bioRxiv. 2024. Update in: Genome Biol. 2024 Oct 10;25(1):265. doi: 10.1186/s13059-024-03409-1 PMID: 38529499 Free PMC article. Updated. Preprint.
Similar articles
-
Phasing Diploid Genome Assembly Graphs with Single-Cell Strand Sequencing.bioRxiv [Preprint]. 2024 Jun 20:2024.02.15.580432. doi: 10.1101/2024.02.15.580432. bioRxiv. 2024. Update in: Genome Biol. 2024 Oct 10;25(1):265. doi: 10.1186/s13059-024-03409-1 PMID: 38529499 Free PMC article. Updated. Preprint.
-
Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm.Nat Methods. 2021 Feb;18(2):170-175. doi: 10.1038/s41592-020-01056-5. Epub 2021 Feb 1. Nat Methods. 2021. PMID: 33526886 Free PMC article.
-
phasebook: haplotype-aware de novo assembly of diploid genomes from long reads.Genome Biol. 2021 Oct 27;22(1):299. doi: 10.1186/s13059-021-02512-x. Genome Biol. 2021. PMID: 34706745 Free PMC article.
-
Haplotyping-Assisted Diploid Assembly and Variant Detection with Linked Reads.Methods Mol Biol. 2023;2590:161-182. doi: 10.1007/978-1-0716-2819-5_11. Methods Mol Biol. 2023. PMID: 36335499 Review.
-
De novo phasing resolves haplotype sequences in complex plant genomes.Plant Biotechnol J. 2022 Jun;20(6):1031-1041. doi: 10.1111/pbi.13815. Epub 2022 Apr 9. Plant Biotechnol J. 2022. PMID: 35332665 Free PMC article. Review.
Cited by
-
Complex genetic variation in nearly complete human genomes.bioRxiv [Preprint]. 2024 Sep 25:2024.09.24.614721. doi: 10.1101/2024.09.24.614721. bioRxiv. 2024. PMID: 39372794 Free PMC article. Preprint.
-
A familial, telomere-to-telomere reference for human de novo mutation and recombination from a four-generation pedigree.bioRxiv [Preprint]. 2024 Aug 5:2024.08.05.606142. doi: 10.1101/2024.08.05.606142. bioRxiv. 2024. PMID: 39149261 Free PMC article. Preprint.
References
-
- Leitwein M, Duranton M, Rougemont Q, Gagnaire P-A, Bernatchez L. Using haplotype information for conservation genomics. Trends Ecol Evol. 2020;35:245–58. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
