

Integrated stress response (ISR) activation and apoptosis through HRI kinase by PG3 and other p53 pathway-restoring cancer therapeutics
- PMID: 39288289
- PMCID: PMC11407758
- DOI: 10.18632/oncotarget.28637
Integrated stress response (ISR) activation and apoptosis through HRI kinase by PG3 and other p53 pathway-restoring cancer therapeutics
Abstract
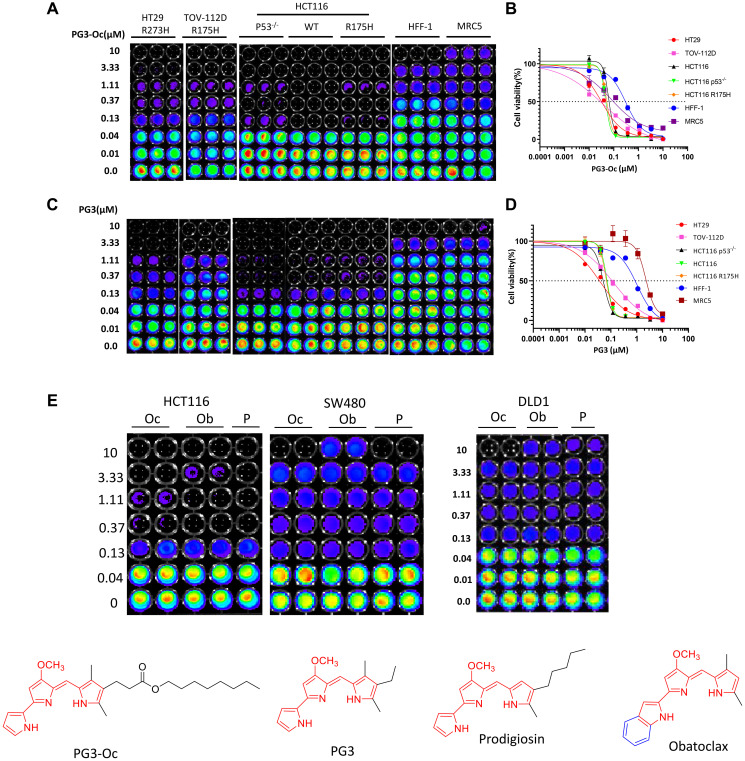
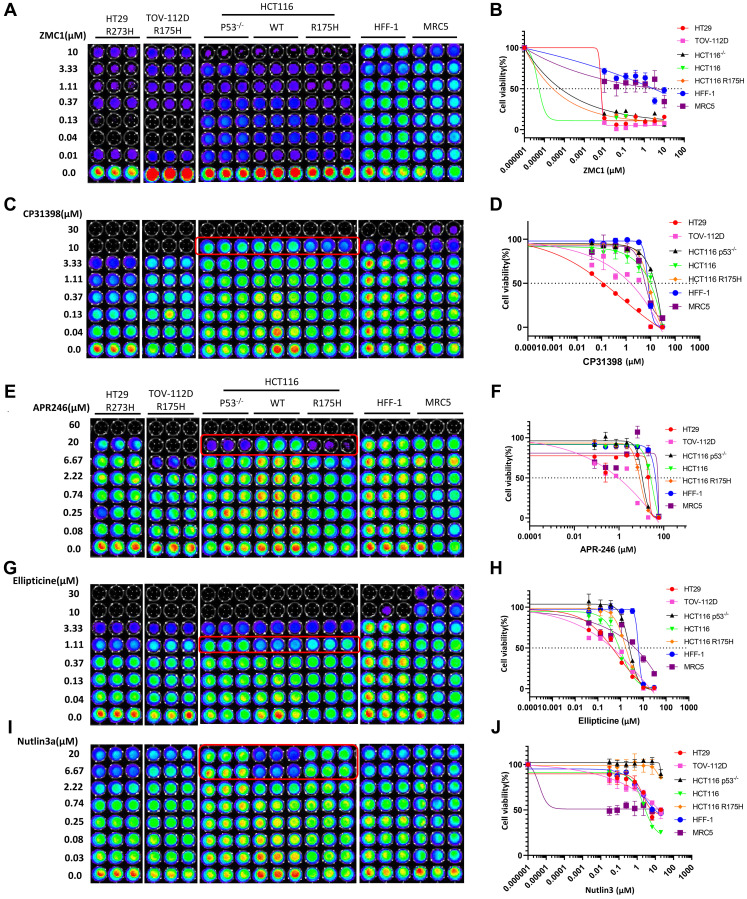
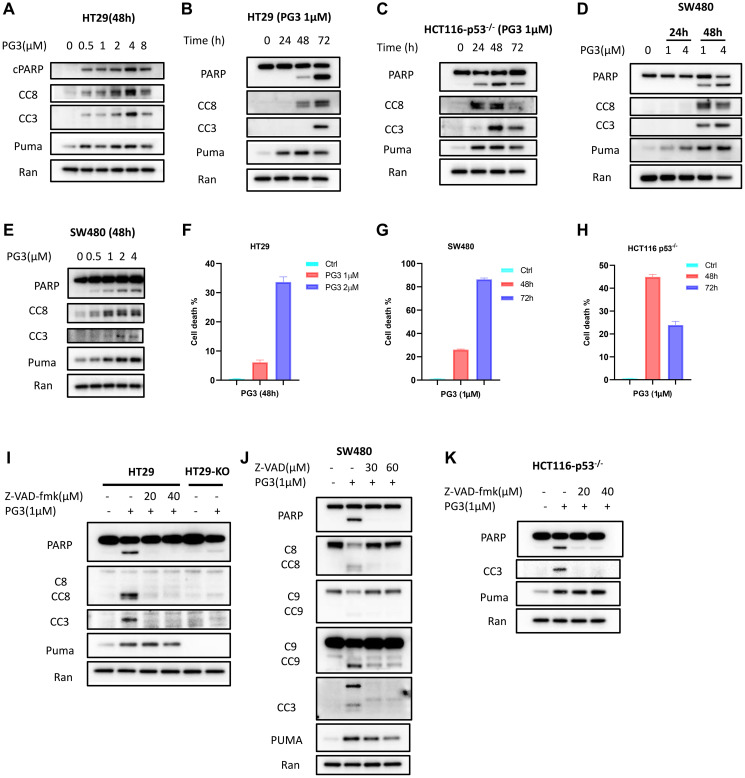
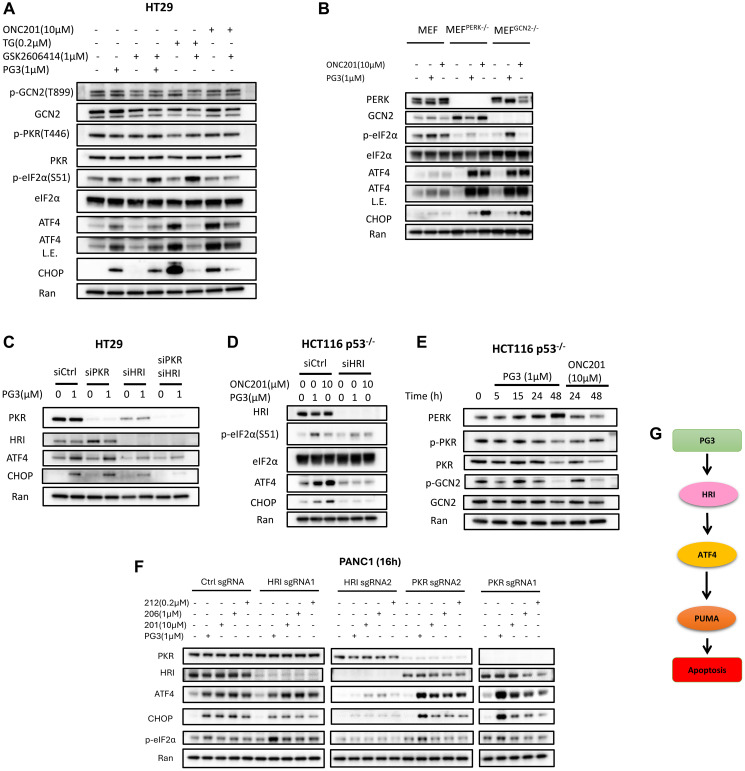
Restoration of the p53 pathway has been a long-term goal in the field of cancer research to treat tumors with mutated p53 and aggressive clinical behavior. p53 pathway restoration in p53-deficient cancers can be achieved by small molecules via p53-dependent or p53-independent processes. Hereafter p53-independent restoration of p53-pathway-signaling in p53-deficient/mutated tumors is referred to as 'restoration of the p53 pathway'. We compare activation of p53 target genes by novel compounds PG3 and PG3-Oc, that activate p53-target genes in a p53-independent manner, and four mutant p53-activating compounds while Nutlin-3a is used as negative control. PG3 and PG3-Oc upregulate p21, PUMA, and DR5 in five cancer cell lines with various p53 mutational statuses through ATF4 (Activating Transcriptional Factor 4) and integrated stress response (ISR) independent of p53. Mutant p53-targeting compounds induce expression of the 3 major downstream p53 target genes and ATF4 in a highly variable and cell-type-dependent manner. PG3 treatment activates ATF4 through ISR via kinase HRI (Heme-Regulated Inhibitor). ATF4 mediates upregulation of PUMA, p21, and NAG-1/GDF15 (Nonsteroidal anti-inflammatory drug-activated gene 1). We note that PUMA mediates apoptosis through activation of caspase-8 in HT29 cells and potentially caspase-10 in SW480 cells. We provide a novel mechanism engaged by PG3 to induce cell death via the HRI/ATF4/PUMA axis. Our results provide unique insights into the mechanism of action of PG3 as a novel cancer therapeutic targeting p53 pathway-like tumor suppression.
Keywords: ATF4; ClpP; HRI; integrated stress response (ISR); mutant p53.
Conflict of interest statement
W.S.E-D. is a co-founder of Oncoceutics, Inc., a subsidiary of Chimerix, p53-Therapeutics, Inc., and SMURF-Therapeutics, Inc. Dr. El-Deiry has disclosed his relationships with Oncoceutics/Chimerix, p53-Therapeutics, Inc., and SMURF-Therapeutics and potential conflicts of interest to his academic institution/employer and is fully compliant with NIH and institutional policy that is managing this potential conflict of interest.
Figures




Similar articles
-
P53-independent partial restoration of the p53 pathway in tumors with mutated p53 through ATF4 transcriptional modulation by ERK1/2 and CDK9.Neoplasia. 2021 Mar;23(3):304-325. doi: 10.1016/j.neo.2021.01.004. Epub 2021 Feb 11. Neoplasia. 2021. PMID: 33582407 Free PMC article.
-
Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA.J Neurosci. 2010 Dec 15;30(50):16938-48. doi: 10.1523/JNEUROSCI.1598-10.2010. J Neurosci. 2010. PMID: 21159964 Free PMC article.
-
Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: evidence for p53-transcription-dependent and -independent pathways.Cancer Biol Ther. 2010 Sep 15;10(6):567-78. doi: 10.4161/cbt.10.6.12535. Epub 2010 Oct 1. Cancer Biol Ther. 2010. PMID: 20595817 Free PMC article.
-
Roles of TP53 in determining therapeutic sensitivity, growth, cellular senescence, invasion and metastasis.Adv Biol Regul. 2017 Jan;63:32-48. doi: 10.1016/j.jbior.2016.10.001. Epub 2016 Oct 6. Adv Biol Regul. 2017. PMID: 27776972 Review.
-
Targeting apoptotic pathways for cancer therapy.J Clin Invest. 2024 Jul 15;134(14):e179570. doi: 10.1172/JCI179570. J Clin Invest. 2024. PMID: 39007268 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
