

From Crypts to Cancer: A Holistic Perspective on Colorectal Carcinogenesis and Therapeutic Strategies
- PMID: 39273409
- PMCID: PMC11395697
- DOI: 10.3390/ijms25179463
From Crypts to Cancer: A Holistic Perspective on Colorectal Carcinogenesis and Therapeutic Strategies
Abstract
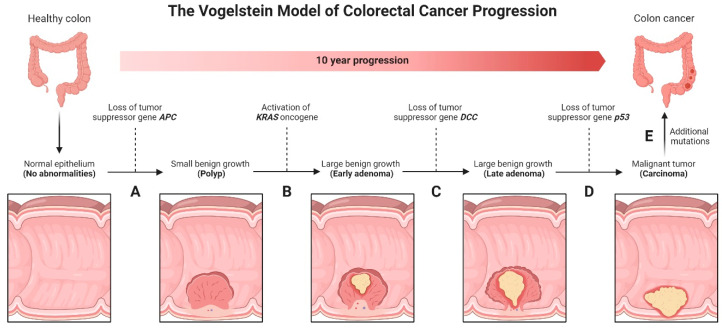
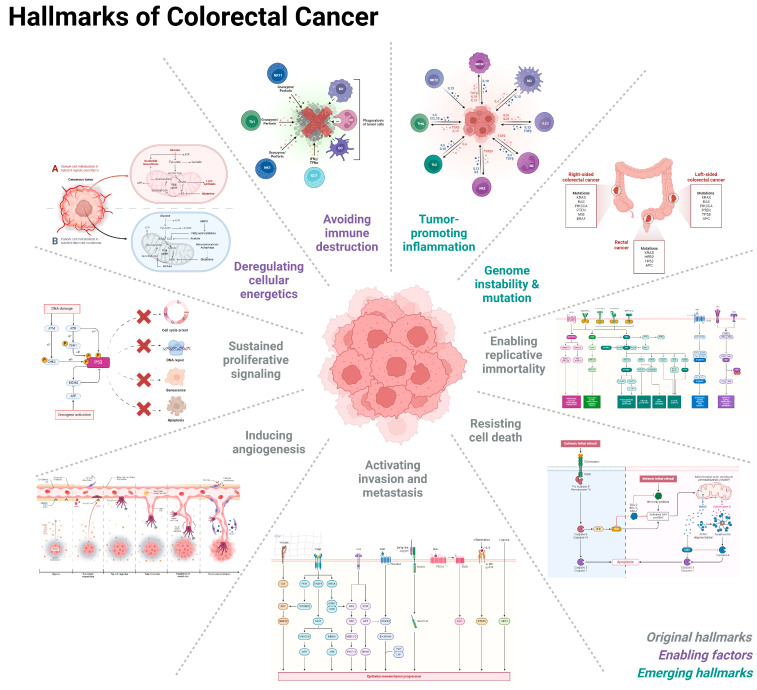
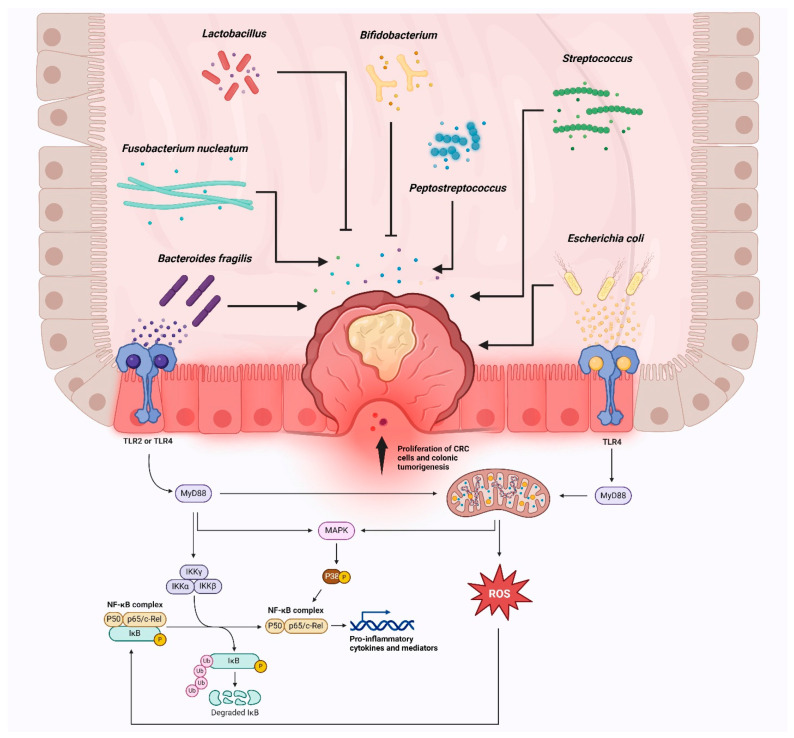
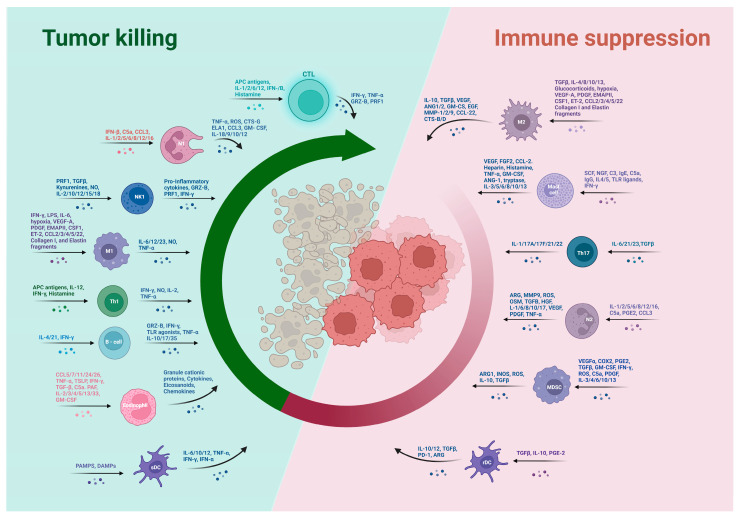
Colorectal cancer (CRC) represents a significant global health burden, with high incidence and mortality rates worldwide. Recent progress in research highlights the distinct clinical and molecular characteristics of colon versus rectal cancers, underscoring tumor location's importance in treatment approaches. This article provides a comprehensive review of our current understanding of CRC epidemiology, risk factors, molecular pathogenesis, and management strategies. We also present the intricate cellular architecture of colonic crypts and their roles in intestinal homeostasis. Colorectal carcinogenesis multistep processes are also described, covering the conventional adenoma-carcinoma sequence, alternative serrated pathways, and the influential Vogelstein model, which proposes sequential APC, KRAS, and TP53 alterations as drivers. The consensus molecular CRC subtypes (CMS1-CMS4) are examined, shedding light on disease heterogeneity and personalized therapy implications.
Keywords: colorectal cancer; epidemiology; molecular pathogenesis; risk factor; therapeutic strategy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Molecular features of colorectal hyperplastic polyps and sessile serrated adenoma/polyps from Korea.Am J Surg Pathol. 2011 Sep;35(9):1274-86. doi: 10.1097/PAS.0b013e318224cd2e. Am J Surg Pathol. 2011. PMID: 21836485
-
Functional links between the microbiome and the molecular pathways of colorectal carcinogenesis.Cancer Metastasis Rev. 2024 Dec;43(4):1463-1474. doi: 10.1007/s10555-024-10215-5. Epub 2024 Sep 28. Cancer Metastasis Rev. 2024. PMID: 39340753 Free PMC article. Review.
-
Molecular pathological classification of colorectal cancer.Virchows Arch. 2016 Aug;469(2):125-34. doi: 10.1007/s00428-016-1956-3. Epub 2016 Jun 20. Virchows Arch. 2016. PMID: 27325016 Free PMC article. Review.
-
Molecular Subtypes of Colorectal Cancer and Their Clinicopathologic Features, With an Emphasis on the Serrated Neoplasia Pathway.Arch Pathol Lab Med. 2016 May;140(5):406-12. doi: 10.5858/arpa.2015-0310-RA. Arch Pathol Lab Med. 2016. PMID: 27128298 Review.
-
Serrated Colorectal Cancer: The Road Less Travelled?Trends Cancer. 2019 Nov;5(11):742-754. doi: 10.1016/j.trecan.2019.09.004. Epub 2019 Oct 28. Trends Cancer. 2019. PMID: 31735291 Free PMC article. Review.
References
-
- Morgan E., Arnold M., Gini A., Lorenzoni V., Cabasag C., Laversanne M., Vignat J., Ferlay J., Murphy N., Bray F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut. 2023;72:338–344. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
