

Inherited human RelB deficiency impairs innate and adaptive immunity to infection
- PMID: 39231201
- PMCID: PMC11406260
- DOI: 10.1073/pnas.2321794121
Inherited human RelB deficiency impairs innate and adaptive immunity to infection
Abstract
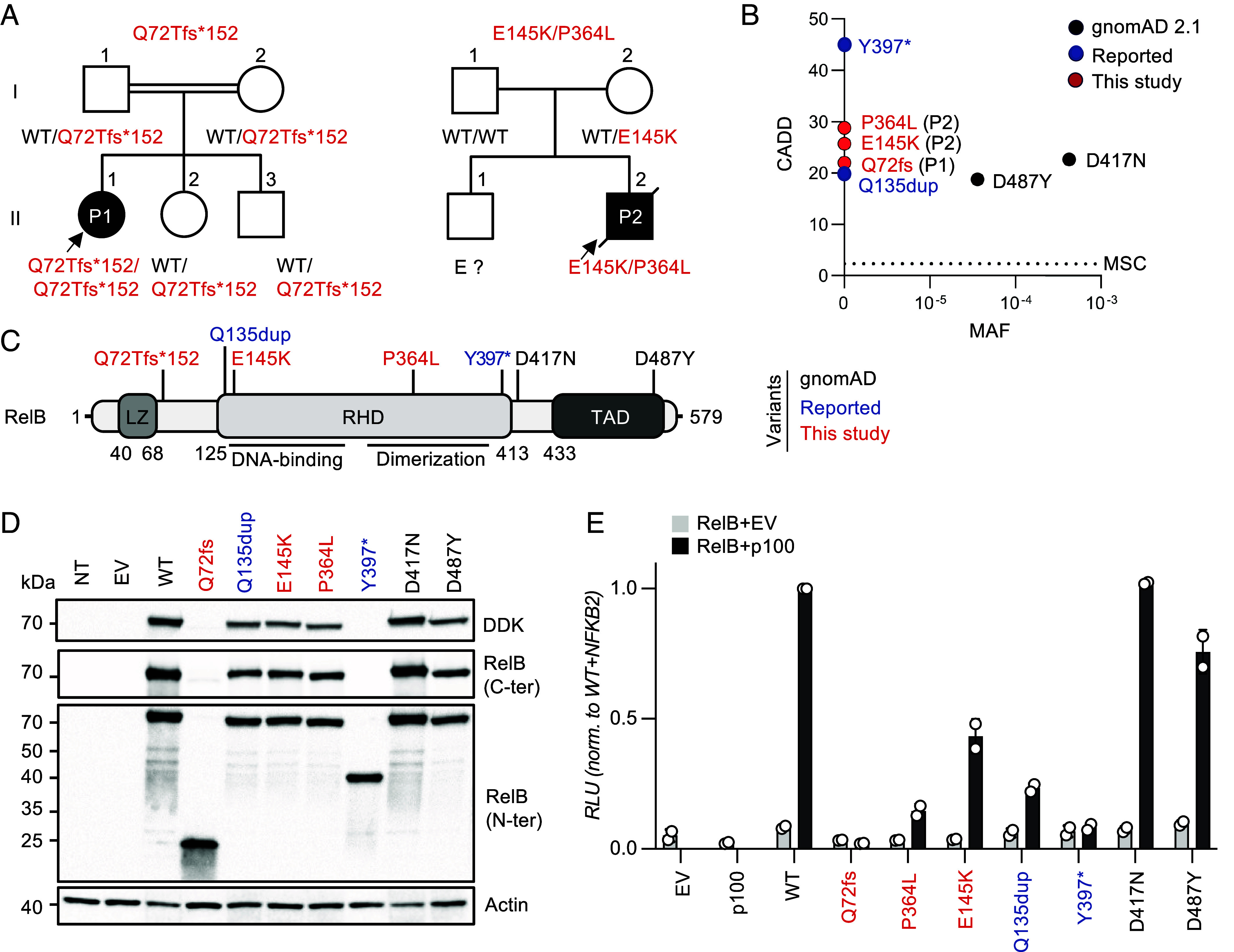
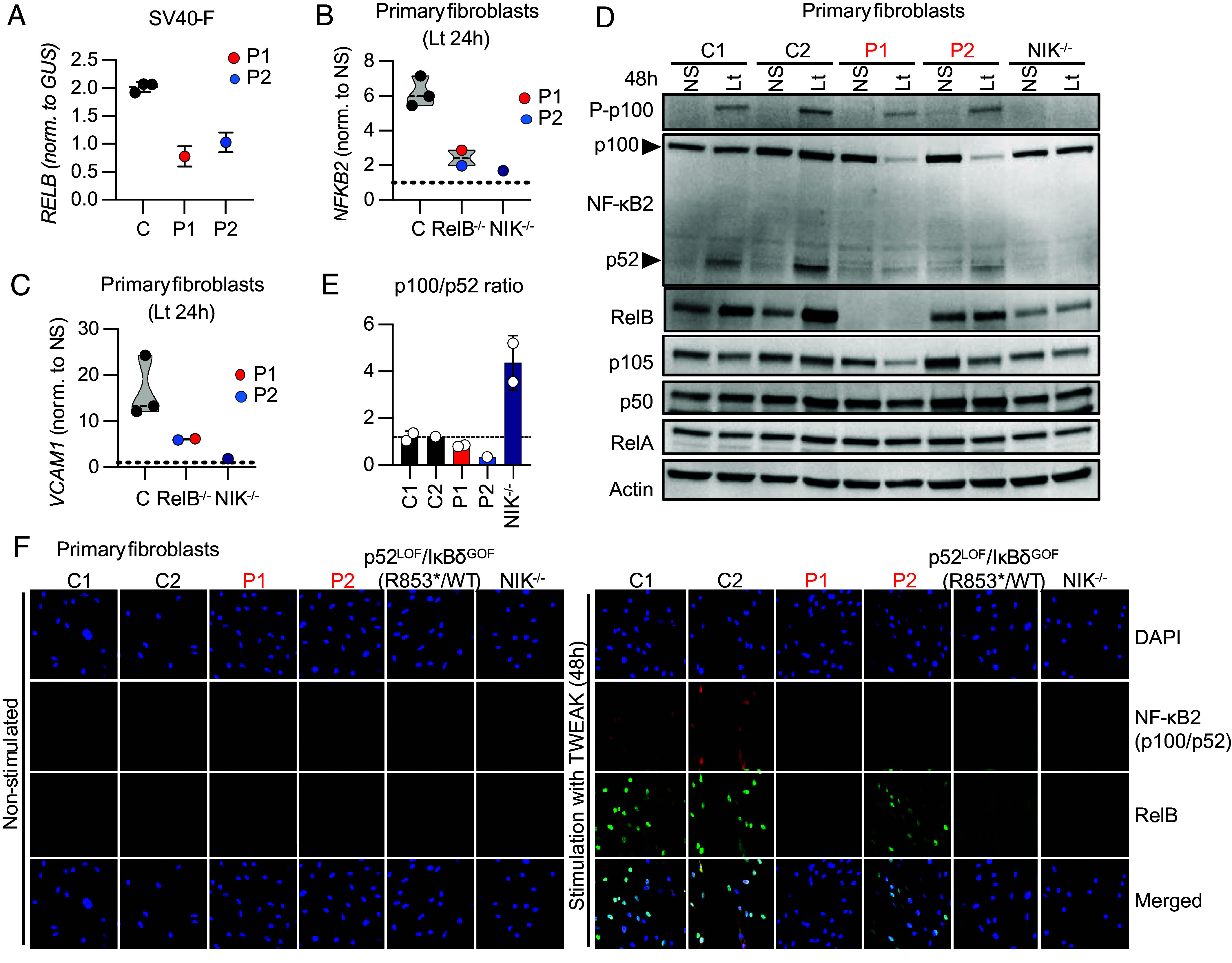
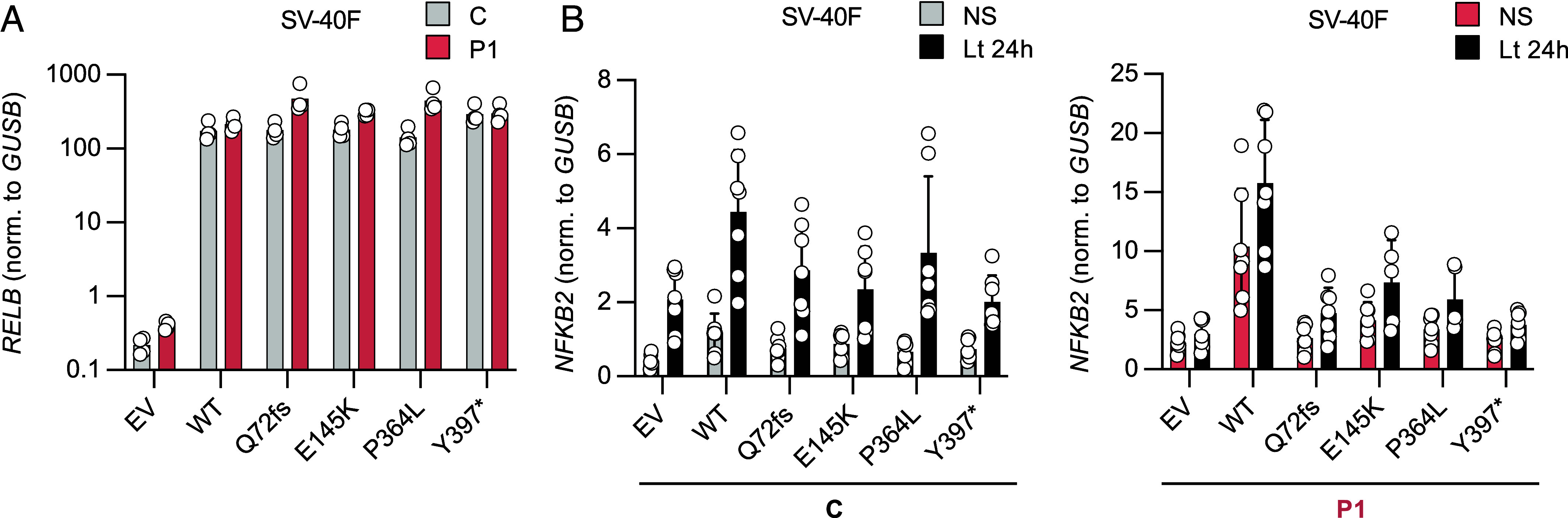
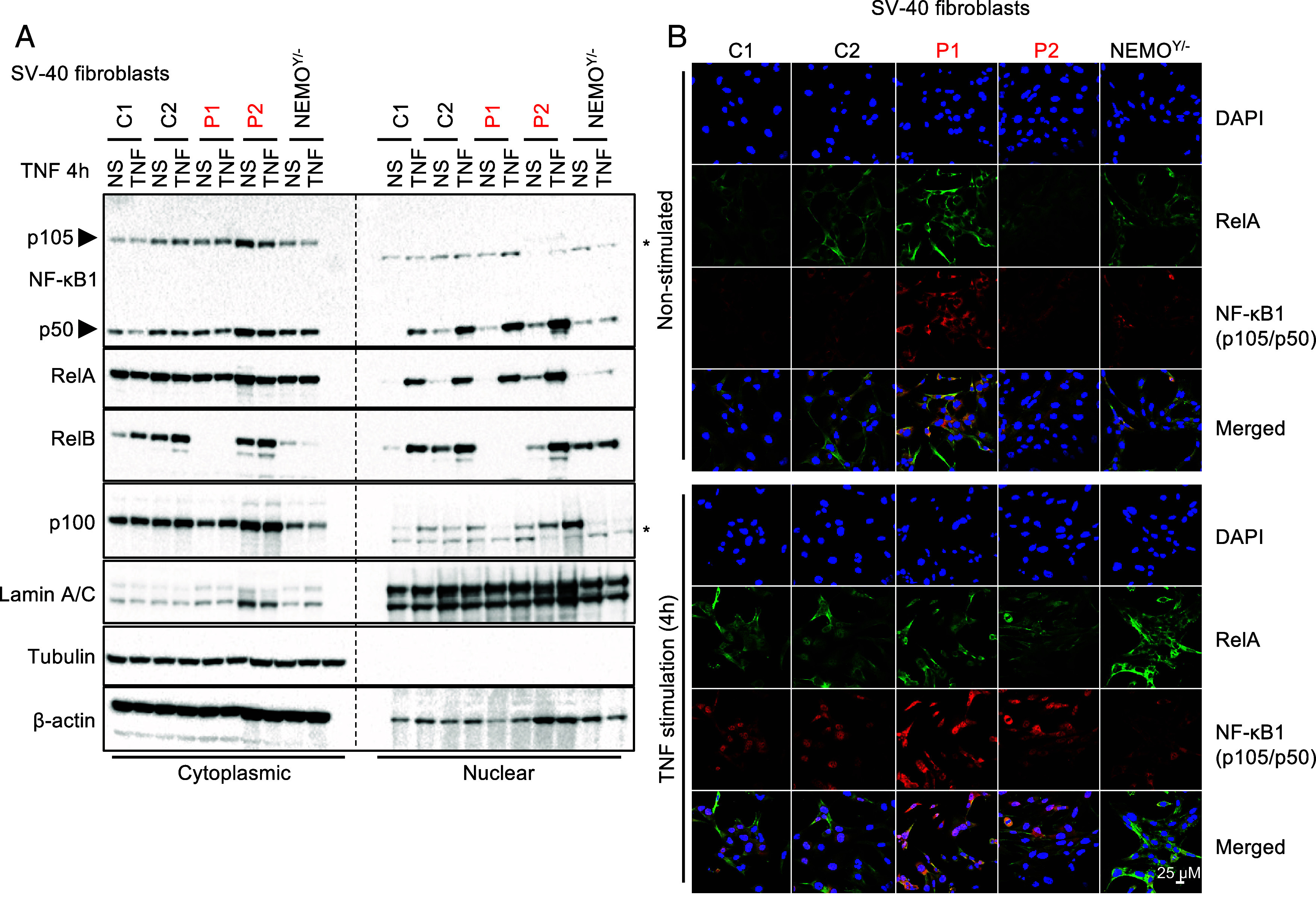
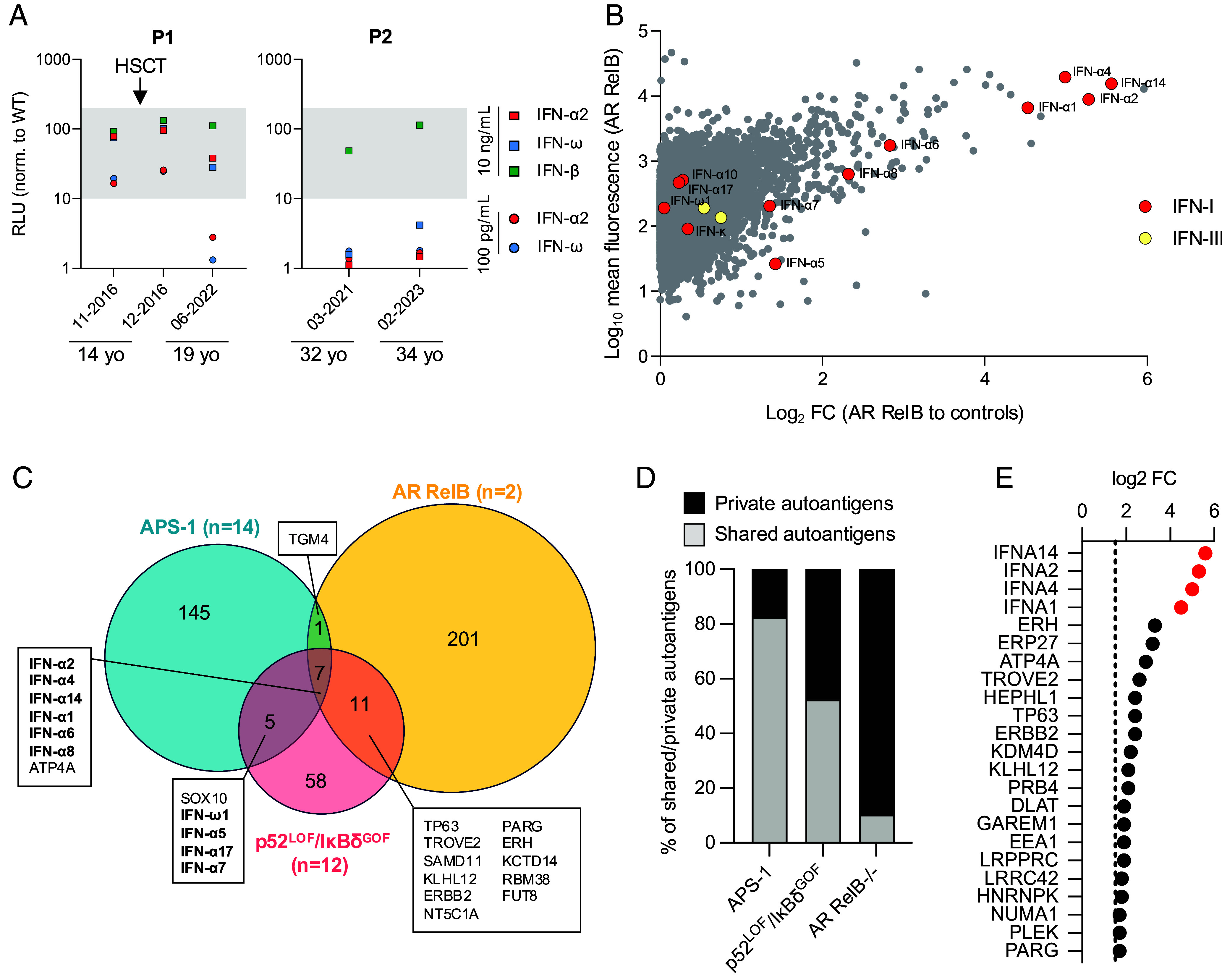
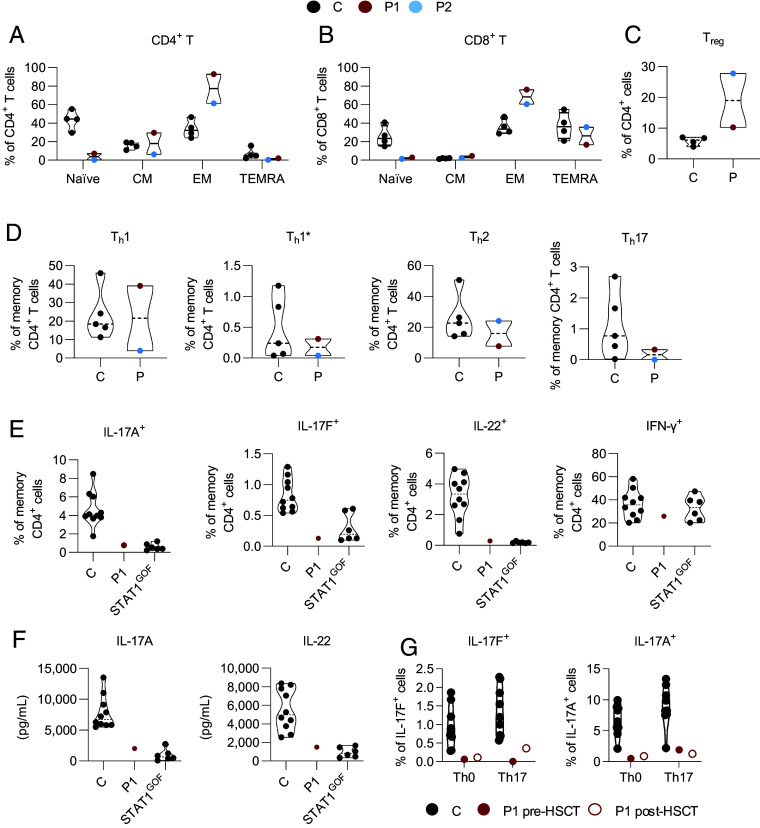
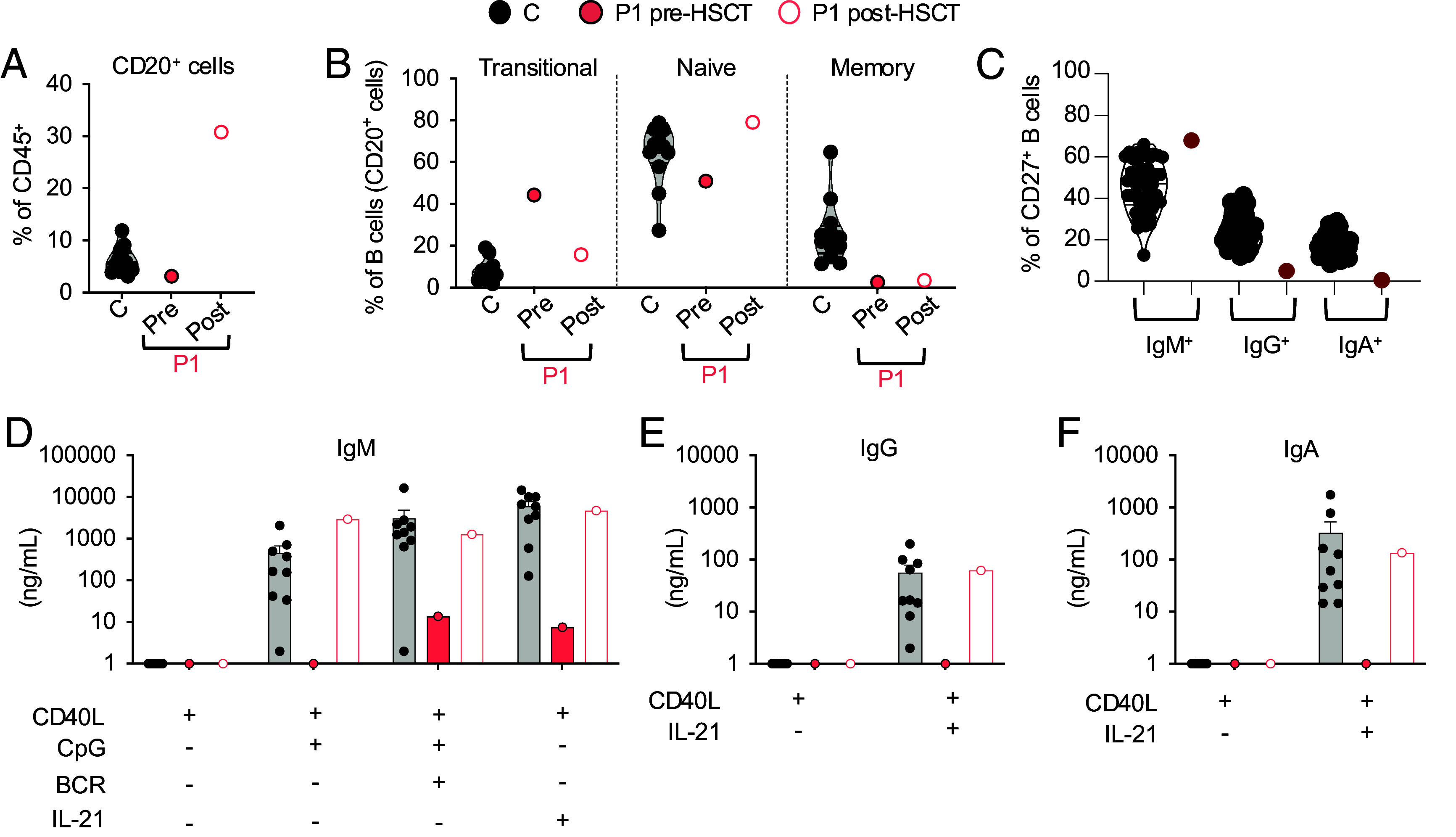
We report two unrelated adults with homozygous (P1) or compound heterozygous (P2) private loss-of-function variants of V-Rel Reticuloendotheliosis Viral Oncogene Homolog B (RELB). The resulting deficiency of functional RelB impairs the induction of NFKB2 mRNA and NF-κB2 (p100/p52) protein by lymphotoxin in the fibroblasts of the patients. These defects are rescued by transduction with wild-type RELB complementary DNA (cDNA). By contrast, the response of RelB-deficient fibroblasts to Tumor Necrosis Factor (TNF) or IL-1β via the canonical NF-κB pathway remains intact. P1 and P2 have low proportions of naïve CD4+ and CD8+ T cells and of memory B cells. Moreover, their naïve B cells cannot differentiate into immunoglobulin G (IgG)- or immunoglobulin A (IgA)-secreting cells in response to CD40L/IL-21, and the development of IL-17A/F-producing T cells is strongly impaired in vitro. Finally, the patients produce neutralizing autoantibodies against type I interferons (IFNs), even after hematopoietic stem cell transplantation, attesting to a persistent dysfunction of thymic epithelial cells in T cell selection and central tolerance to some autoantigens. Thus, inherited human RelB deficiency disrupts the alternative NF-κB pathway, underlying a T- and B cell immunodeficiency, which, together with neutralizing autoantibodies against type I IFNs, confers a predisposition to viral, bacterial, and fungal infections.
Keywords: NF-κB pathway; RelB deficiency; autoantibodies; immunodeficiency; type I IFNs.
Conflict of interest statement
Competing interests statement:Tim Niehues was a co-author on publications with the following co-authors within the last 4 y: K.B., C.S.M., and S.G.T. (https://doi.org/10.1182/blood.2020006738) and K.B. (https://doi.org/10.1016/j.jaci.2019.11.051; https://doi.org/10.1002/eji.202048713). Andrew R. Gennery was a co-author with S.G.T. (https://doi.org/10.1016/j.jaci.2020.09.010; https://doi.org/10.1016/j.jaci.2022.09.002) within the last 4 y.
Figures
Similar articles
-
Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency.Nature. 2023 Nov;623(7988):803-813. doi: 10.1038/s41586-023-06717-x. Epub 2023 Nov 8. Nature. 2023. PMID: 37938781 Free PMC article.
-
Immune Differentiation Regulator p100 Tunes NF-κB Responses to TNF.Front Immunol. 2019 May 7;10:997. doi: 10.3389/fimmu.2019.00997. eCollection 2019. Front Immunol. 2019. PMID: 31134075 Free PMC article.
-
Impairment of Mature B Cell Maintenance upon Combined Deletion of the Alternative NF-κB Transcription Factors RELB and NF-κB2 in B Cells.J Immunol. 2016 Mar 15;196(6):2591-601. doi: 10.4049/jimmunol.1501120. Epub 2016 Feb 5. J Immunol. 2016. PMID: 26851215 Free PMC article.
-
The Alternative NF-κB Pathway in Regulatory T Cell Homeostasis and Suppressive Function.J Immunol. 2018 Apr 1;200(7):2362-2371. doi: 10.4049/jimmunol.1800042. Epub 2018 Feb 19. J Immunol. 2018. PMID: 29459403 Free PMC article.
-
p100 Deficiency is insufficient for full activation of the alternative NF-κB pathway: TNF cooperates with p52-RelB in target gene transcription.PLoS One. 2012;7(8):e42741. doi: 10.1371/journal.pone.0042741. Epub 2012 Aug 6. PLoS One. 2012. PMID: 22880094 Free PMC article.
References
-
- Fusco A. J., et al. , The NF-κB subunit RelB controls p100 processing by competing with the kinases NIK and IKK1 for binding to p100. Sci. Signal. 9, ra96 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- N/A/Department of Health of the New South Wales Government of Australia
- N/A/Bettencourt-Schueller Foundation
- ANR-20-CO11-0001/Agence Nationale de la Recherche (ANR)
- N/A/Imagine Institute
- N/A/HHMI (HHMI)
- N/A/Stavros Niarchos Foundation (SNF)
- UL1TR001866/HHS | NIH | National Center for Advancing Translational Sciences (NCATS)
- N/A/HHS | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- 1042925/DHAC | National Health and Medical Research Council (NHMRC)
- F99 CA274708/CA/NCI NIH HHS/United States
- N/A/Fondation du Souffle (FdS)
- EQU201903007798/Fondation pour la Recherche Médicale (FRM)
- N/A/apanese Foundation for Pediatric Research
- 1176665/DHAC | National Health and Medical Research Council (NHMRC)
- N/A/William E. Ford, General Atlantic's Chairman and Chief Executive Officer, Gabriel Caillaux, General Atlantic's Co-President, Managing Director and Head of business in EMEA, General Atlantic Foundation
- R01 AI127564/AI/NIAID NIH HHS/United States
- ANR-21-RHUS-08-COVIFERON/Agence Nationale de la Recherche (ANR)
- R01AI127564/HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- N/A/Grandir - Fonds de solidarité pour l'enfance
- N/A/European Society for Immunodeficiencies (ESID)
- N/A/Rockefeller University (The Rockefeller Institute)
- ANR-22-CE15-0046/Agence Nationale de la Recherche (ANR)
- 01057100 (UNDINE)/EC | HORIZON EUROPE Framework Programme (Horizon Europe)
- F99CA274708/HHS | NIH | National Cancer Institute (NCI)
- N/A/New York Hideyo Noguchi Memorial Society
- UL1 TR001866/TR/NCATS NIH HHS/United States
- 1113904/DHAC | National Health and Medical Research Council (NHMRC)
- ANR-18-CE93-0008/Agence Nationale de la Recherche (ANR)
- N/A/Honjo International Scholarship Foundation (HISF)
- N/A/FULBRIGHT | Fulbright U.S. Scholar Program
- N/A/Paris Cite University
- ANR-10-LABX-62-IBEID/Agence Nationale de la Recherche (ANR)
- N/A/Philipp Foundation
- ANR-10-IAHU-01/Agence Nationale de la Recherche (ANR)
- N/A/HHS | NIH | NIAID | Division of Intramural Research (DIR, NIAID)
- N/A/Institut National de la Santé et de la Recherche Médicale (Inserm)
- N/A/Square Foundation
- N/A/Funai Foundation for Information Technology
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
