

GGTyper: genotyping complex structural variants using short-read sequencing data
- PMID: 39230689
- PMCID: PMC11373317
- DOI: 10.1093/bioinformatics/btae391
GGTyper: genotyping complex structural variants using short-read sequencing data
Abstract
Motivation: Complex structural variants (SVs) are genomic rearrangements that involve multiple segments of DNA. They contribute to human diversity and have been shown to cause Mendelian disease. Nevertheless, our abilities to analyse complex SVs are very limited. As opposed to deletions and other canonical types of SVs, there are no established tools that have explicitly been designed for analysing complex SVs.
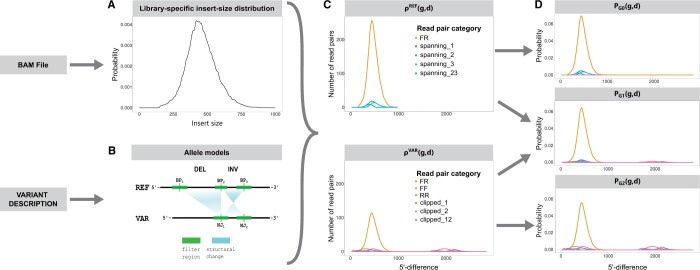
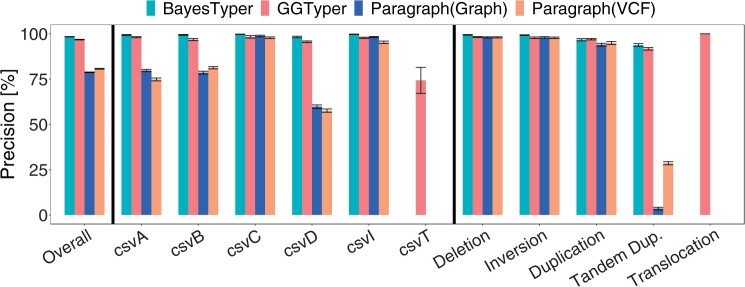
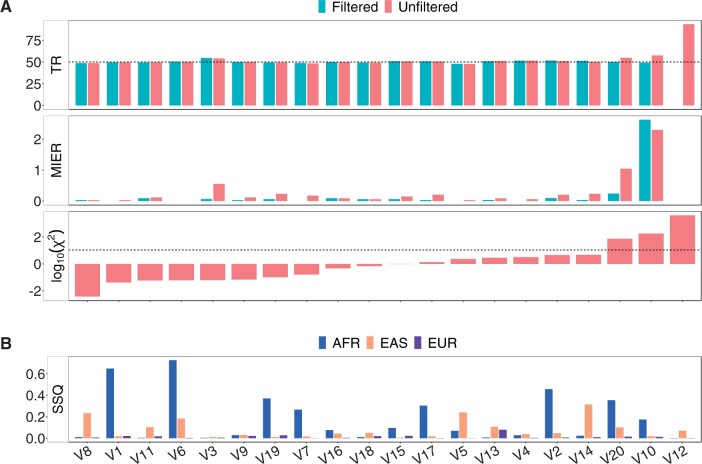
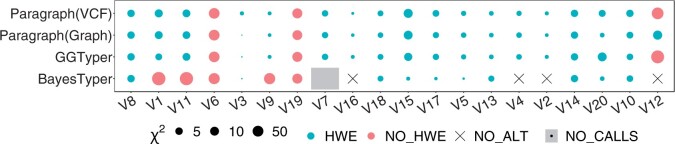
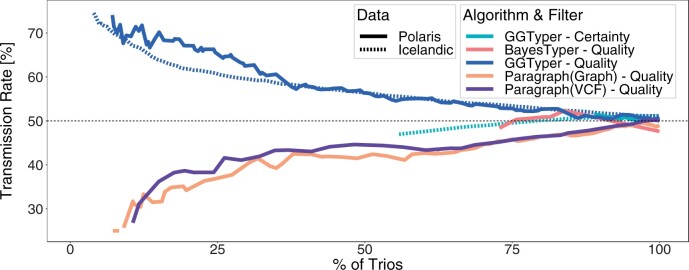
Results: Here, we describe a new computational approach that we specifically designed for genotyping complex SVs in short-read sequenced genomes. Given a variant description, our approach computes genotype-specific probability distributions for observing aligned read pairs with a wide range of properties. Subsequently, these distributions can be used to efficiently determine the most likely genotype for any set of aligned read pairs observed in a sequenced genome. In addition, we use these distributions to compute a genotyping difficulty for a given variant, which predicts the amount of data needed to achieve a reliable call. Careful evaluation confirms that our approach outperforms other genotypers by making reliable genotype predictions across both simulated and real data. On up to 7829 human genomes, we achieve high concordance with population-genetic assumptions and expected inheritance patterns. On simulated data, we show that precision correlates well with our prediction of genotyping difficulty. This together with low memory and time requirements makes our approach well-suited for application in biomedical studies involving small to very large numbers of short-read sequenced genomes.
Availability and implementation: Source code is available at https://github.com/kehrlab/Complex-SV-Genotyping.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
B.V.H. is an employee of deCODE genetics/Amgen Inc.
Figures

Similar articles
-
NPSV-deep: a deep learning method for genotyping structural variants in short read genome sequencing data.Bioinformatics. 2024 Mar 4;40(3):btae129. doi: 10.1093/bioinformatics/btae129. Bioinformatics. 2024. PMID: 38444093 Free PMC article.
-
SVJedi: genotyping structural variations with long reads.Bioinformatics. 2020 Nov 1;36(17):4568-4575. doi: 10.1093/bioinformatics/btaa527. Bioinformatics. 2020. PMID: 32437523
-
Discovery and genotyping of novel sequence insertions in many sequenced individuals.Bioinformatics. 2017 Jul 15;33(14):i161-i169. doi: 10.1093/bioinformatics/btx254. Bioinformatics. 2017. PMID: 28881988 Free PMC article.
-
A decade of structural variants: description, history and methods to detect structural variation.Brief Funct Genomics. 2015 Sep;14(5):305-14. doi: 10.1093/bfgp/elv014. Epub 2015 Apr 15. Brief Funct Genomics. 2015. PMID: 25877305 Review.
-
A Practical Guide for Structural Variation Detection in the Human Genome.Curr Protoc Hum Genet. 2020 Sep;107(1):e103. doi: 10.1002/cphg.103. Curr Protoc Hum Genet. 2020. PMID: 32813322 Free PMC article. Review.
References
-
- Beyter D, Ingimundardottir H, Oddsson A et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet 2021;53:779–86. - PubMed
-
- Chen X, Schulz-Trieglaff O, Shaw R et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2015;32:1220–2. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
