

Update on the Genetics of Osteogenesis Imperfecta
- PMID: 39127989
- PMCID: PMC11607015
- DOI: 10.1007/s00223-024-01266-5
Update on the Genetics of Osteogenesis Imperfecta
Abstract
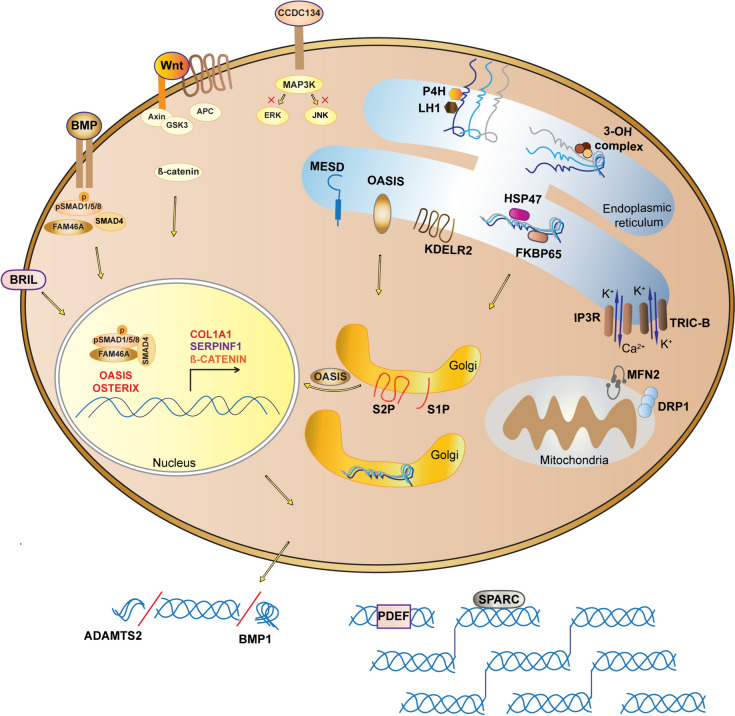
Osteogenesis imperfecta (OI) is a heterogeneous heritable skeletal dysplasia characterized by bone fragility and deformity, growth deficiency, and other secondary connective tissue defects. OI is now understood as a collagen-related disorder caused by defects of genes whose protein products interact with collagen for folding, post-translational modification, processing and trafficking, affecting bone mineralization and osteoblast differentiation. This review provides the latest updates on genetics of OI, including new developments in both dominant and rare OI forms, as well as the signaling pathways involved in OI pathophysiology. There is a special emphasis on discoveries of recessive mutations in TENT5A, MESD, KDELR2 and CCDC134 whose causality of OI types XIX, XX, XXI and XXI, respectively, is now established and expends the complexity of mechanisms underlying OI to overlap LRP5/6 and MAPK/ERK pathways. We also review in detail new discoveries connecting the known OI types to each other, which may underlie an eventual understanding of a final common pathway in OI cellular and bone biology.
Keywords: Bone mineralization; IFITM5/BRIL; MAPK/ERK; Mitochondria; Osteoblast differentiation; Osteogenesis imperfecta; PDEF; RIP/MBTPS2.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Declarations. Conflict of interest: None of the authors has any conflict of interest.
Figures
Similar articles
-
Bone Quality and Mineralization and Effects of Treatment in Osteogenesis Imperfecta.Calcif Tissue Int. 2024 Dec;115(6):777-804. doi: 10.1007/s00223-024-01263-8. Epub 2024 Sep 4. Calcif Tissue Int. 2024. PMID: 39231826 Review.
-
A Dyadic Nosology for Osteogenesis Imperfecta and Bone Fragility Syndromes 2024.Calcif Tissue Int. 2024 Dec;115(6):873-890. doi: 10.1007/s00223-024-01248-7. Epub 2024 Jun 28. Calcif Tissue Int. 2024. PMID: 38942908 Free PMC article. Review.
-
Extra-Skeletal Manifestations in Osteogenesis Imperfecta Mouse Models.Calcif Tissue Int. 2024 Dec;115(6):847-862. doi: 10.1007/s00223-024-01213-4. Epub 2024 Apr 19. Calcif Tissue Int. 2024. PMID: 38641703 Review.
-
Cardiovascular disease in adults with osteogenesis imperfecta: Clinical characteristics, care recommendations and research priorities identified using a modified Delphi technique.J Bone Miner Res. 2024 Dec 12:zjae197. doi: 10.1093/jbmr/zjae197. Online ahead of print. J Bone Miner Res. 2024. PMID: 39665364
-
Lung function in adult patients with osteogenesis imperfecta: a cohort study.Orphanet J Rare Dis. 2024 Dec 4;19(1):455. doi: 10.1186/s13023-024-03452-y. Orphanet J Rare Dis. 2024. PMID: 39627862 Free PMC article.
Cited by
-
Orthopedic Surgery in Osteogenesis Imperfecta in Adults.Calcif Tissue Int. 2024 Dec;115(6):976-988. doi: 10.1007/s00223-024-01306-0. Epub 2024 Nov 16. Calcif Tissue Int. 2024. PMID: 39550451 Review.
-
Osteogenesis Imperfecta from Bench to Bedside and from Cradle to Grave.Calcif Tissue Int. 2024 Dec;115(6):775-776. doi: 10.1007/s00223-024-01304-2. Epub 2024 Nov 20. Calcif Tissue Int. 2024. PMID: 39565402 No abstract available.
References
-
- Marini JC, Cabral WA (2018) Osteogenesis imperfecta. Genet Bone Biol Skeletal Dis, pp 397–420
-
- Raghunath M, Bruckner P, Steinmann B (1994) Delayed triple helix formation of mutant collagen from patient with osteogenesis imperfecta. J Mol Biol 236(3):940–949 - PubMed
-
- Malfait F, Symoens S, De Backer J, Hermanns-Lê T, Sakalihasan N, Lapière CM et al (2007) Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat 28(4):387–395 - PubMed
-
- Cabral WA, Makareeva E, Letocha AD, Scribanu N, Fertala A, Steplewski A, et al (2007) Y‐position cysteine substitution in type I collagen (α1 (I) R888C/p. R1066C) is associated with osteogenesis imperfecta/Ehlers‐Danlos syndrome phenotype. Human Mutation 28(4):396–405 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
