

Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2
- PMID: 39125583
- PMCID: PMC11311688
- DOI: 10.3390/ijms25158012
Potential Transcriptional Enhancers in Coronaviruses: From Infectious Bronchitis Virus to SARS-CoV-2
Abstract
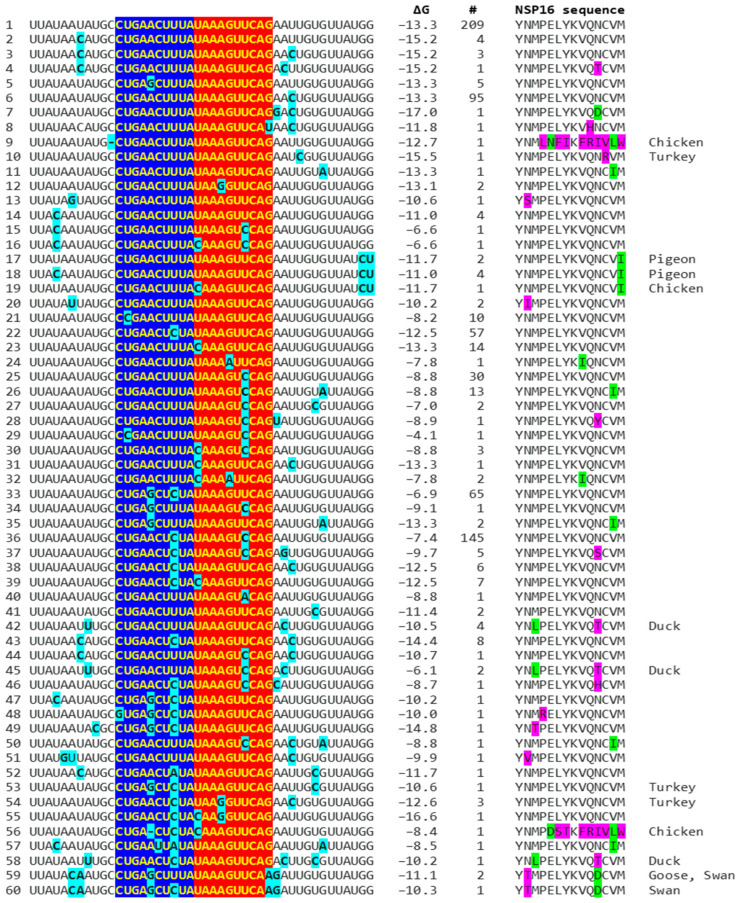
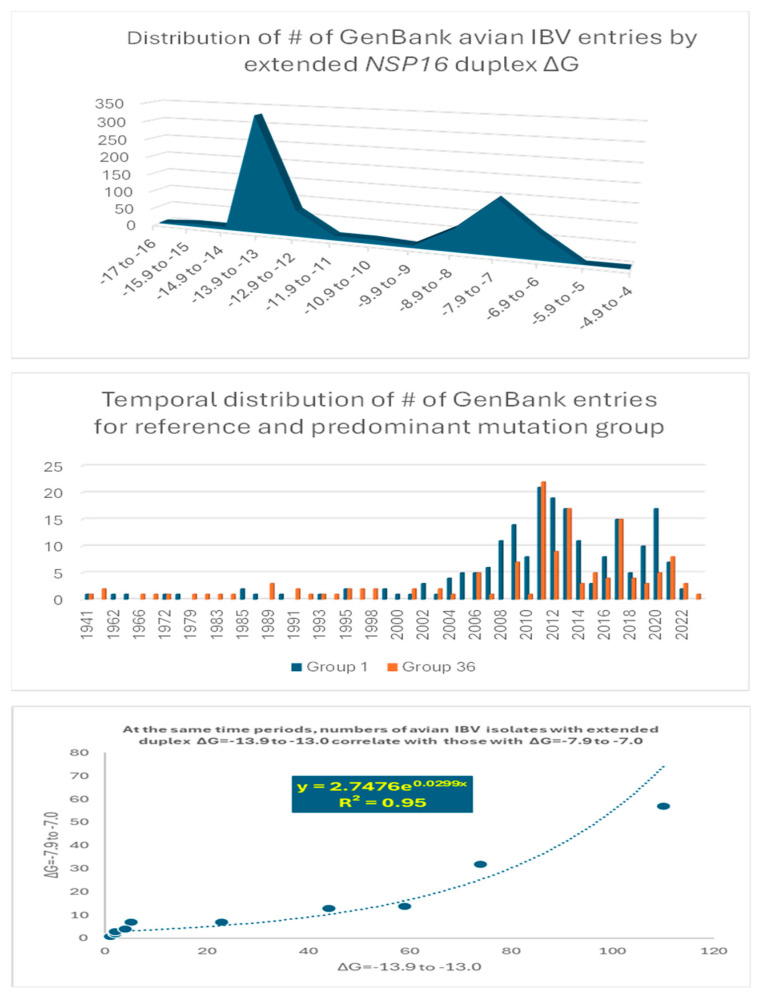
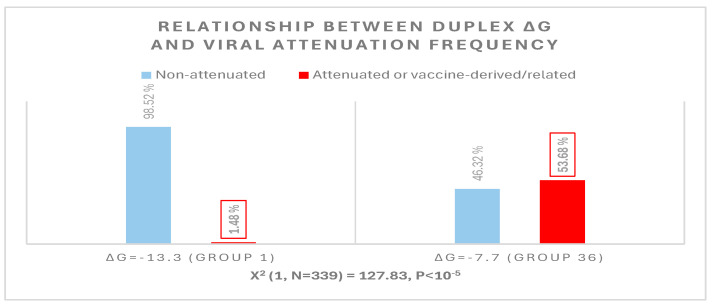
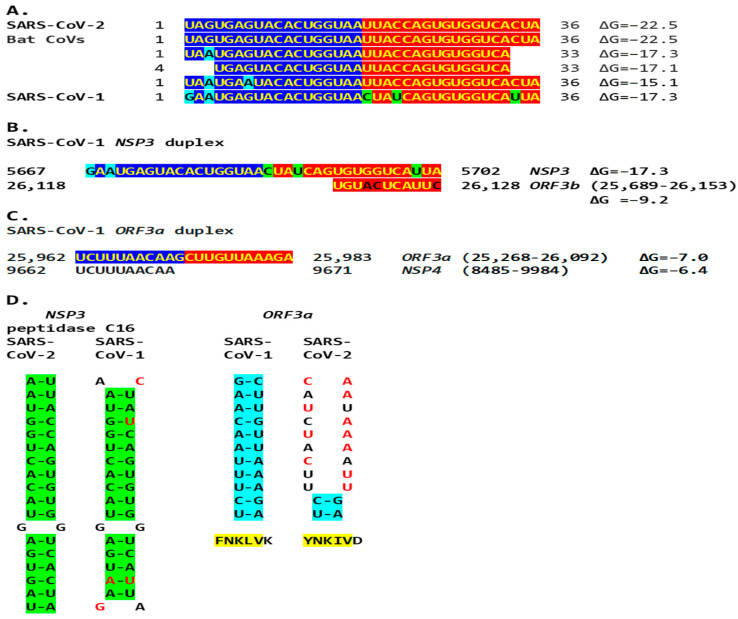
Coronaviruses constitute a global threat to human and animal health. It is essential to investigate the long-distance RNA-RNA interactions that approximate remote regulatory elements in strategies, including genome circularization, discontinuous transcription, and transcriptional enhancers, aimed at the rapid replication of their large genomes, pathogenicity, and immune evasion. Based on the primary sequences and modeled RNA-RNA interactions of two experimentally defined coronaviral enhancers, we detected via an in silico primary and secondary structural analysis potential enhancers in various coronaviruses, from the phylogenetically ancient avian infectious bronchitis virus (IBV) to the recently emerged SARS-CoV-2. These potential enhancers possess a core duplex-forming region that could transition between closed and open states, as molecular switches directed by viral or host factors. The duplex open state would pair with remote sequences in the viral genome and modulate the expression of downstream crucial genes involved in viral replication and host immune evasion. Consistently, variations in the predicted IBV enhancer region or its distant targets coincide with cases of viral attenuation, possibly driven by decreased open reading frame (ORF)3a immune evasion protein expression. If validated experimentally, the annotated enhancer sequences could inform structural prediction tools and antiviral interventions.
Keywords: IBV; SARS-CoV-2; coronavirus; enhancer; host immune evasion; long-range RNA interactions; viral attenuation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures













Similar articles
-
Pervasive RNA Secondary Structure in the Genomes of SARS-CoV-2 and Other Coronaviruses.mBio. 2020 Oct 30;11(6):e01661-20. doi: 10.1128/mBio.01661-20. mBio. 2020. PMID: 33127861 Free PMC article.
-
Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense.Mol Biol Evol. 2020 Sep 1;37(9):2699-2705. doi: 10.1093/molbev/msaa094. Mol Biol Evol. 2020. PMID: 32289821 Free PMC article.
-
Deletion of the s2m RNA Structure in the Avian Coronavirus Infectious Bronchitis Virus and Human Astrovirus Results in Sequence Insertions.J Virol. 2023 Mar 30;97(3):e0003823. doi: 10.1128/jvi.00038-23. Epub 2023 Feb 13. J Virol. 2023. PMID: 36779761 Free PMC article.
-
Properties of Coronavirus and SARS-CoV-2.Malays J Pathol. 2020 Apr;42(1):3-11. Malays J Pathol. 2020. PMID: 32342926 Review.
-
Overview of Immune Response During SARS-CoV-2 Infection: Lessons From the Past.Front Immunol. 2020 Aug 7;11:1949. doi: 10.3389/fimmu.2020.01949. eCollection 2020. Front Immunol. 2020. PMID: 32849654 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
