

Insights into the Gene Expression Profile of Classical Hodgkin Lymphoma: A Study towards Discovery of Novel Therapeutic Targets
- PMID: 39124881
- PMCID: PMC11314437
- DOI: 10.3390/molecules29153476
Insights into the Gene Expression Profile of Classical Hodgkin Lymphoma: A Study towards Discovery of Novel Therapeutic Targets
Abstract
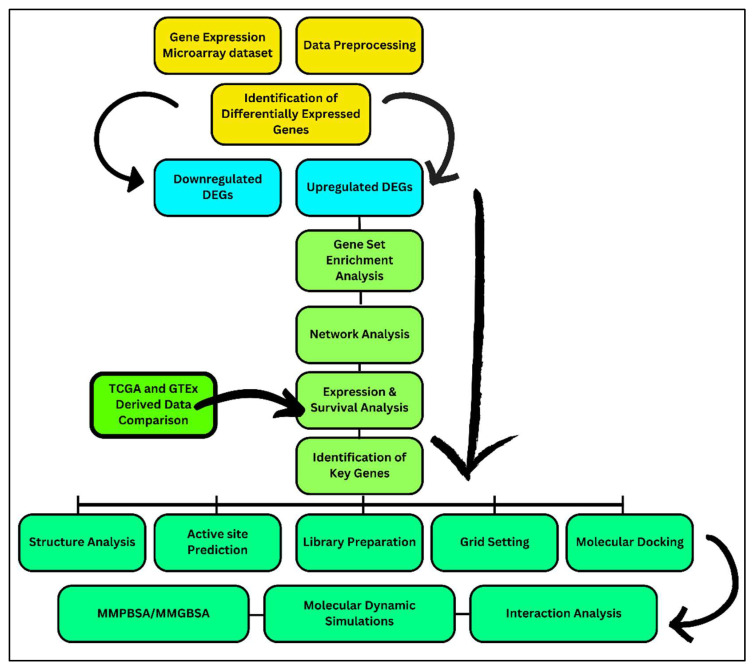
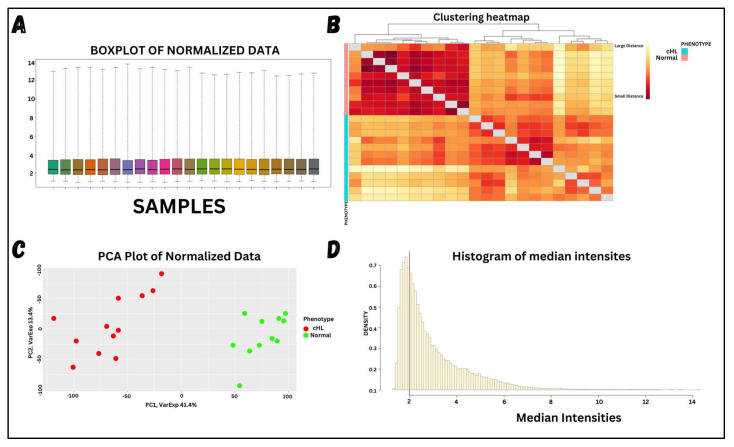
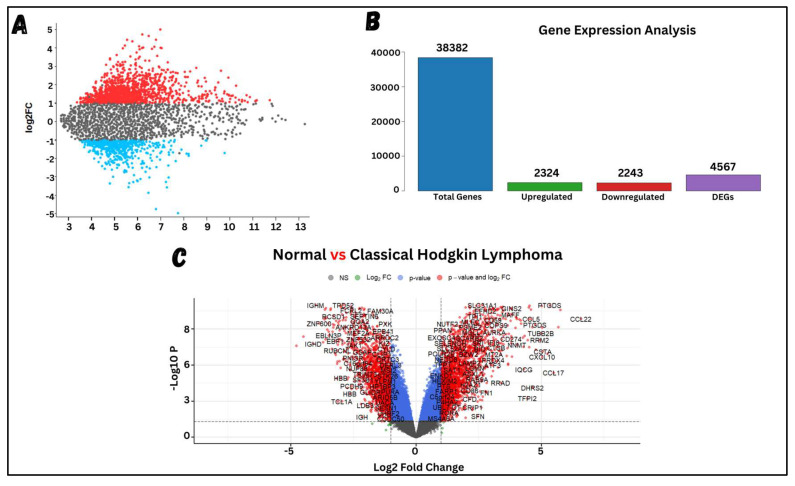
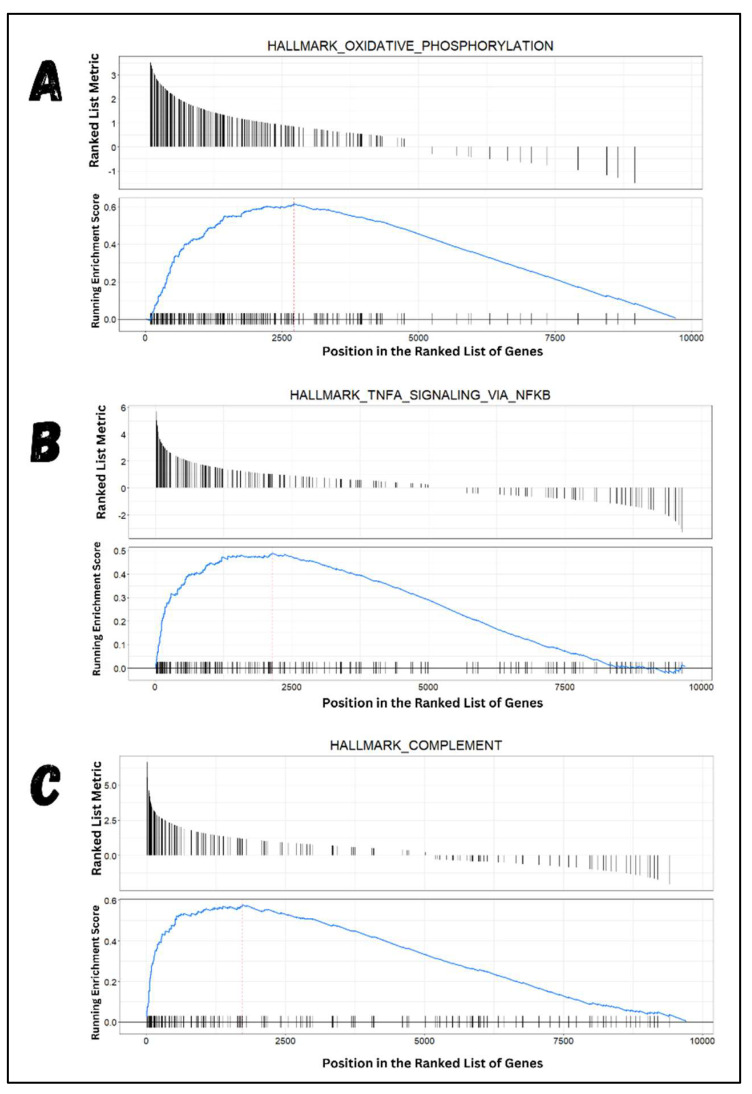
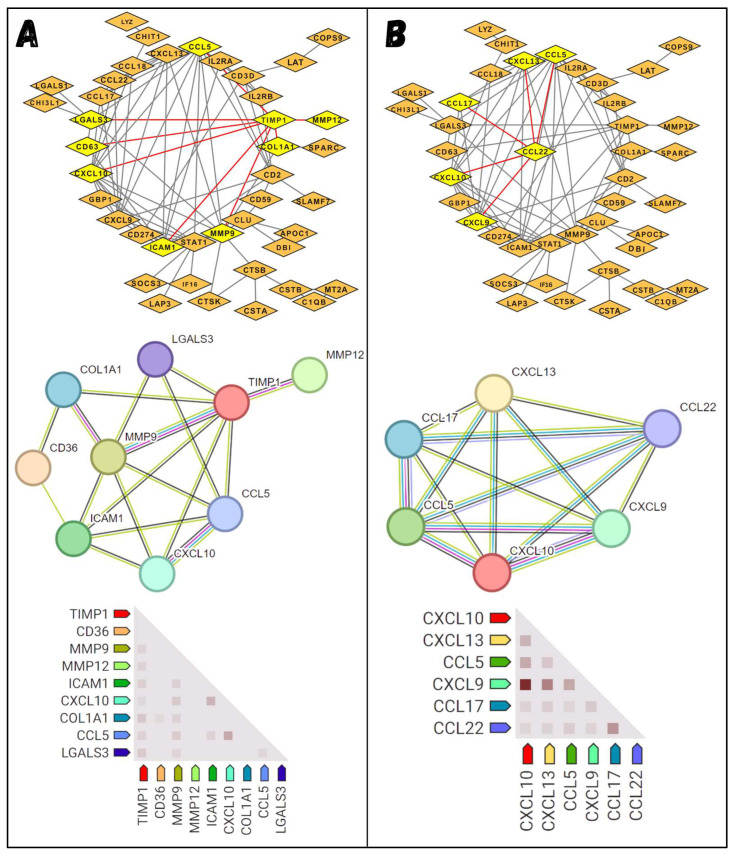
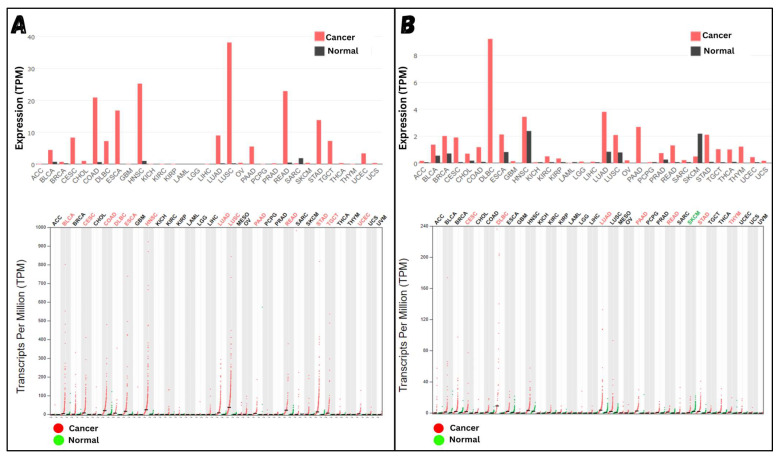
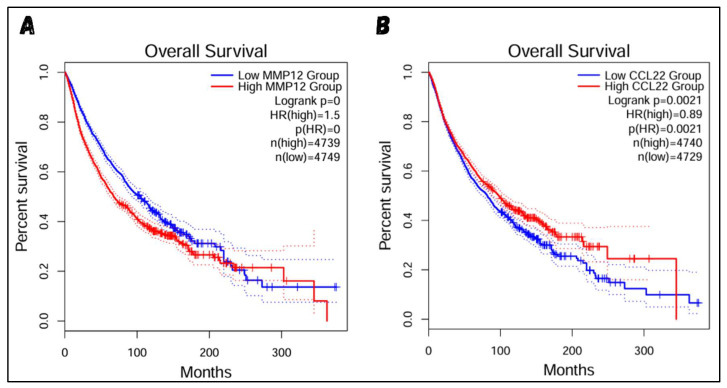
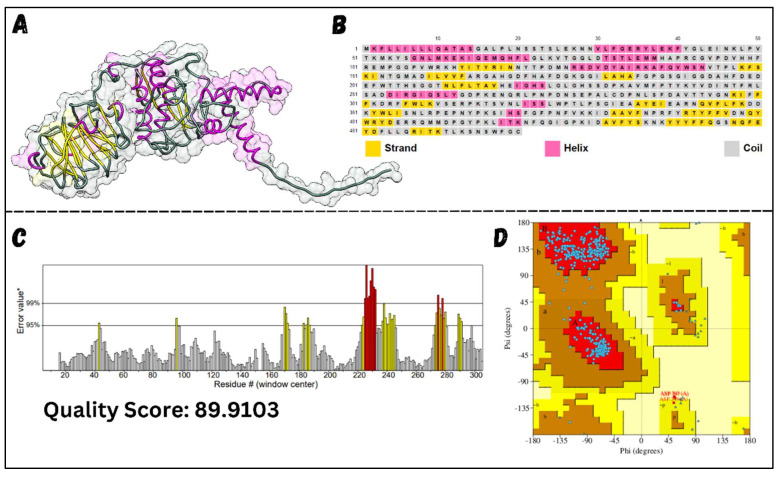
Classical Hodgkin lymphoma (cHL) is a common B-cell cancer and a significant health concern, especially in Western and Asian countries. Despite the effectiveness of chemotherapy, many relapse cases are being reported, highlighting the need for improved treatments. This study aimed to address this issue by discovering biomarkers through the analysis of gene expression data specific to cHL. Additionally, potential anticancer inhibitors were explored to target the discovered biomarkers. This study proceeded by retrieving microarray gene expression data from cHL patients, which was then analyzed to identify significant differentially expressed genes (DEGs). Functional and network annotation of the upregulated genes revealed the active involvement of matrix metallopeptidase 12 (MMP12) and C-C motif metallopeptidase ligand 22 (CCL22) genes in the progression of cHL. Additionally, the mentioned genes were found to be actively involved in cancer-related pathways, i.e., oxidative phosphorylation, complement pathway, myc_targets_v1 pathway, TNFA signaling via NFKB, etc., and showed strong associations with other genes known to promote cancer progression. MMP12, topping the list with a logFC value of +6.6378, was selected for inhibition using docking and simulation strategies. The known anticancer compounds were docked into the active site of the MMP12 molecular structure, revealing significant binding scores of -7.7 kcal/mol and -7.6 kcal/mol for BDC_24037121 and BDC_27854277, respectively. Simulation studies of the docked complexes further supported the effective binding of the ligands, yielding MMGBSA and MMPBSA scores of -78.08 kcal/mol and -82.05 kcal/mol for MMP12-BDC_24037121 and -48.79 kcal/mol and -49.67 kcal/mol for MMP12-BDC_27854277, respectively. Our findings highlight the active role of MMP12 in the progression of cHL, with known compounds effectively inhibiting its function and potentially halting the advancement of cHL. Further exploration of downregulated genes is warranted, as associated genes may play a role in cHL. Additionally, CCL22 should be considered for further investigation due to its significant role in the progression of cHL.
Keywords: cancer therapy; classical Hodgkin lymphoma; docking; drug target; gene expression; microarray data analysis; simulations.
Conflict of interest statement
The author declares no conflicts of interest.
Figures


Similar articles
-
Identification of key genes and pathways associated with classical Hodgkin lymphoma by bioinformatics analysis.Mol Med Rep. 2017 Oct;16(4):4685-4693. doi: 10.3892/mmr.2017.7158. Epub 2017 Aug 3. Mol Med Rep. 2017. PMID: 28791394 Free PMC article.
-
MicroRNA signatures and treatment response in patients with advanced classical Hodgkin lymphoma.Br J Haematol. 2013 Aug;162(3):336-47. doi: 10.1111/bjh.12390. Epub 2013 Jun 1. Br J Haematol. 2013. PMID: 23725219 Free PMC article.
-
Pathway-focused gene expression profiles and immunohistochemistry detection identify contrasting association of caspase 3 (CASP3) expression with prognosis in pediatric classical Hodgkin lymphoma.Hematol Oncol. 2018 Oct;36(4):663-670. doi: 10.1002/hon.2523. Epub 2018 Jun 14. Hematol Oncol. 2018. PMID: 29901224
-
A chemoinformatic-biophysics based approach to identify novel anti-virulent compounds against Pseudomonas aeruginosa disulfide-bond protein A1.J Biomol Struct Dyn. 2023 Aug 7:1-10. doi: 10.1080/07391102.2023.2245470. Online ahead of print. J Biomol Struct Dyn. 2023. PMID: 37551016 Review.
-
Epigenetic abnormalities of classical Hodgkin lymphoma and its effect on immune escape.Cell Biochem Funct. 2020 Apr;38(3):242-248. doi: 10.1002/cbf.3463. Epub 2019 Nov 11. Cell Biochem Funct. 2020. PMID: 31709594 Review.
References
-
- Achkova D. Development of Immunotherapy for Classical Hodgkin’s Lymphoma and Anaplastic Large Cell Lymphoma Using Colony-Stimulating Factor-1 Receptor Re-Targeted T-Lymphocytes. Guy’s, King’s and St. Thomas’s School of Medicine; London, UK: 2016.
-
- Liu D. Tumors and Cancers. CRC Press; Boca Raton, FL, USA: 2017. Hodgkin Lymphoma; pp. 171–176.
-
- Gao J., Chen Y., Wu P., Wang F., Tao H., Shen Q., Wang S., Gong S., Zhang X., Zhou Z., et al. Causes of Death and Effect of Non-Cancer-Specific Death on Rates of Overall Survival in Adult Classic Hodgkin Lymphoma: A Populated-Based Competing Risk Analysis. BMC Cancer. 2021;21:955. doi: 10.1186/s12885-021-08683-x. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
