

Insights into early animal evolution from the genome of the xenacoelomorph worm Xenoturbella bocki
- PMID: 39109482
- PMCID: PMC11521371
- DOI: 10.7554/eLife.94948
Insights into early animal evolution from the genome of the xenacoelomorph worm Xenoturbella bocki
Abstract
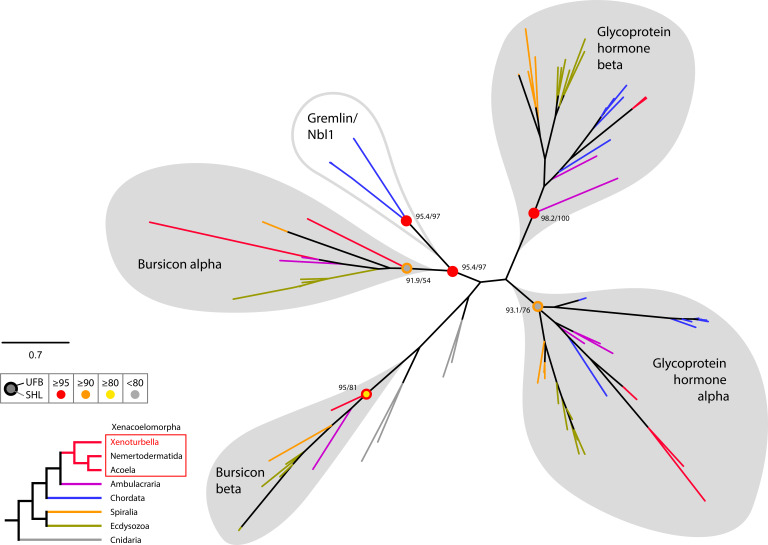
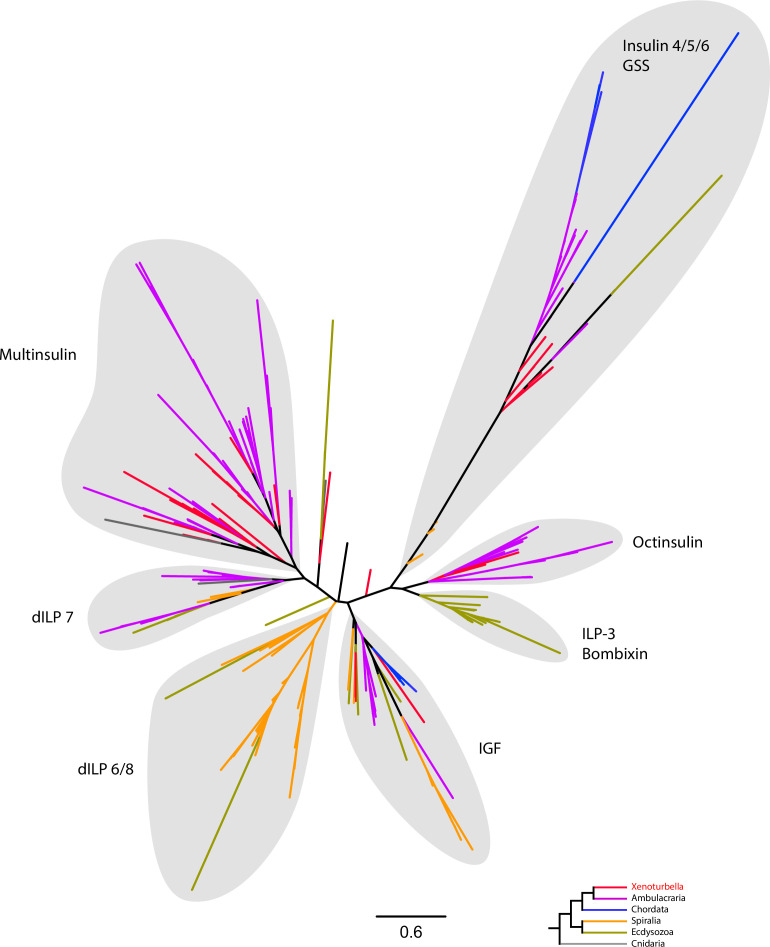
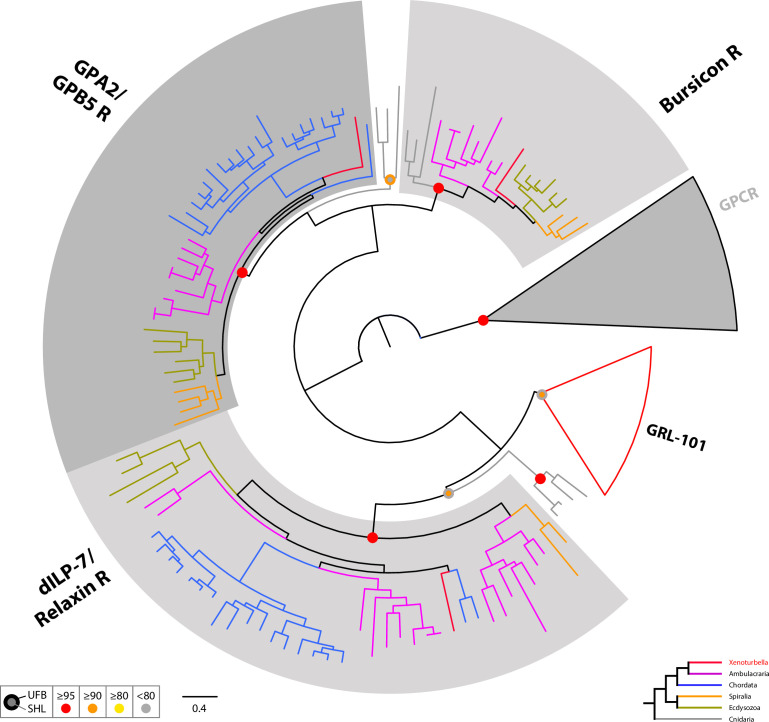
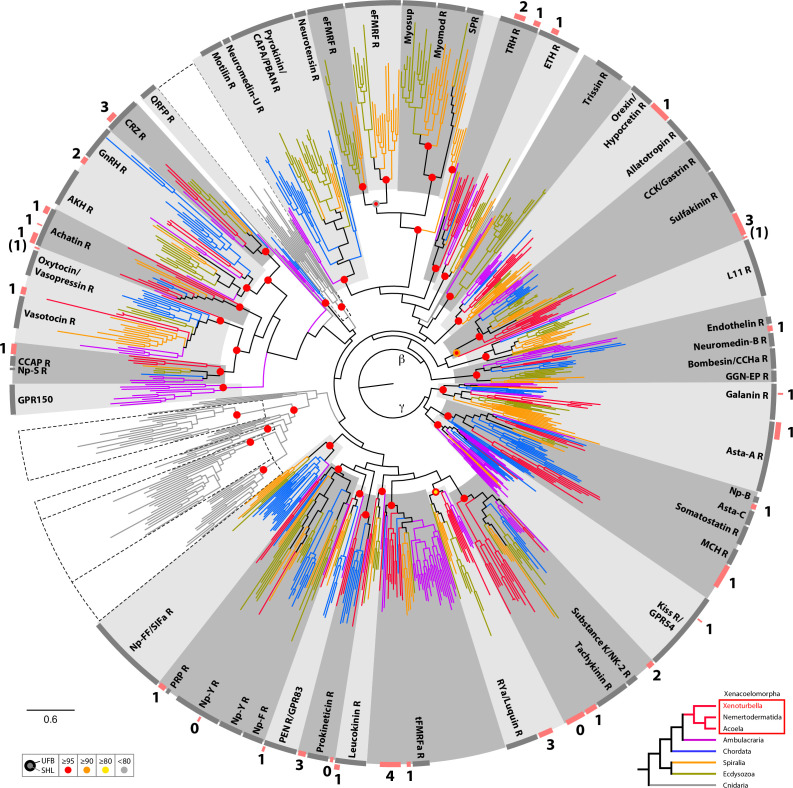
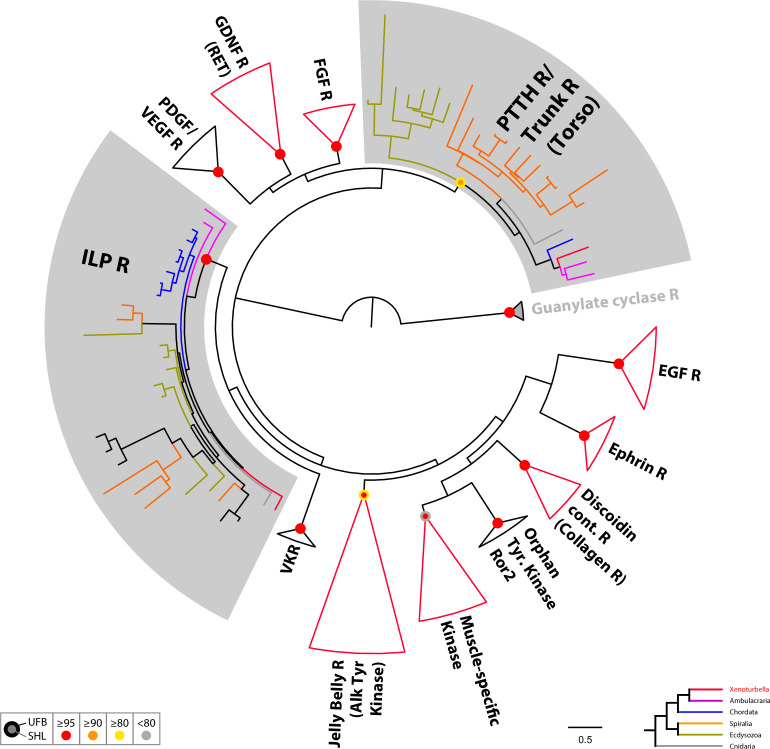
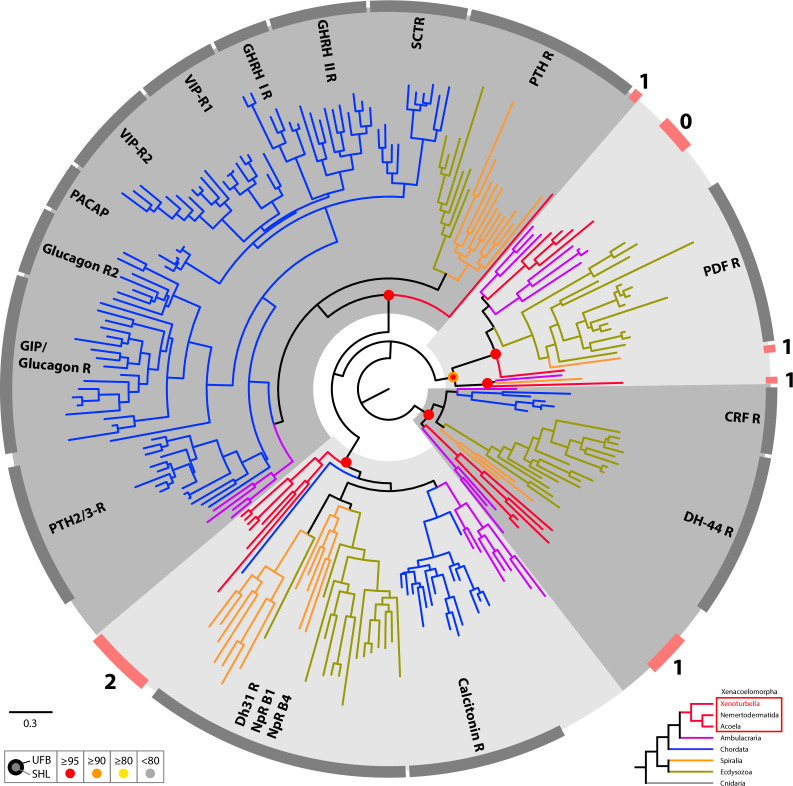
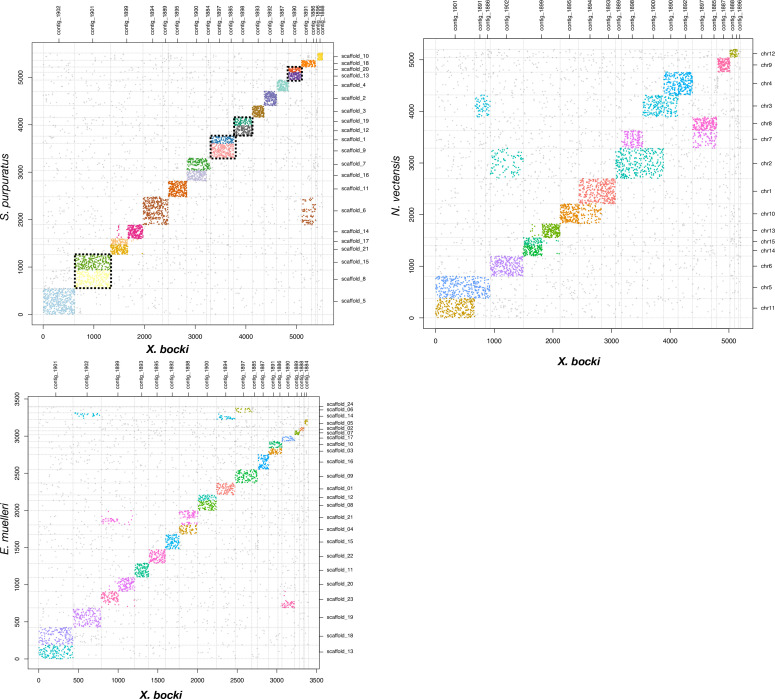
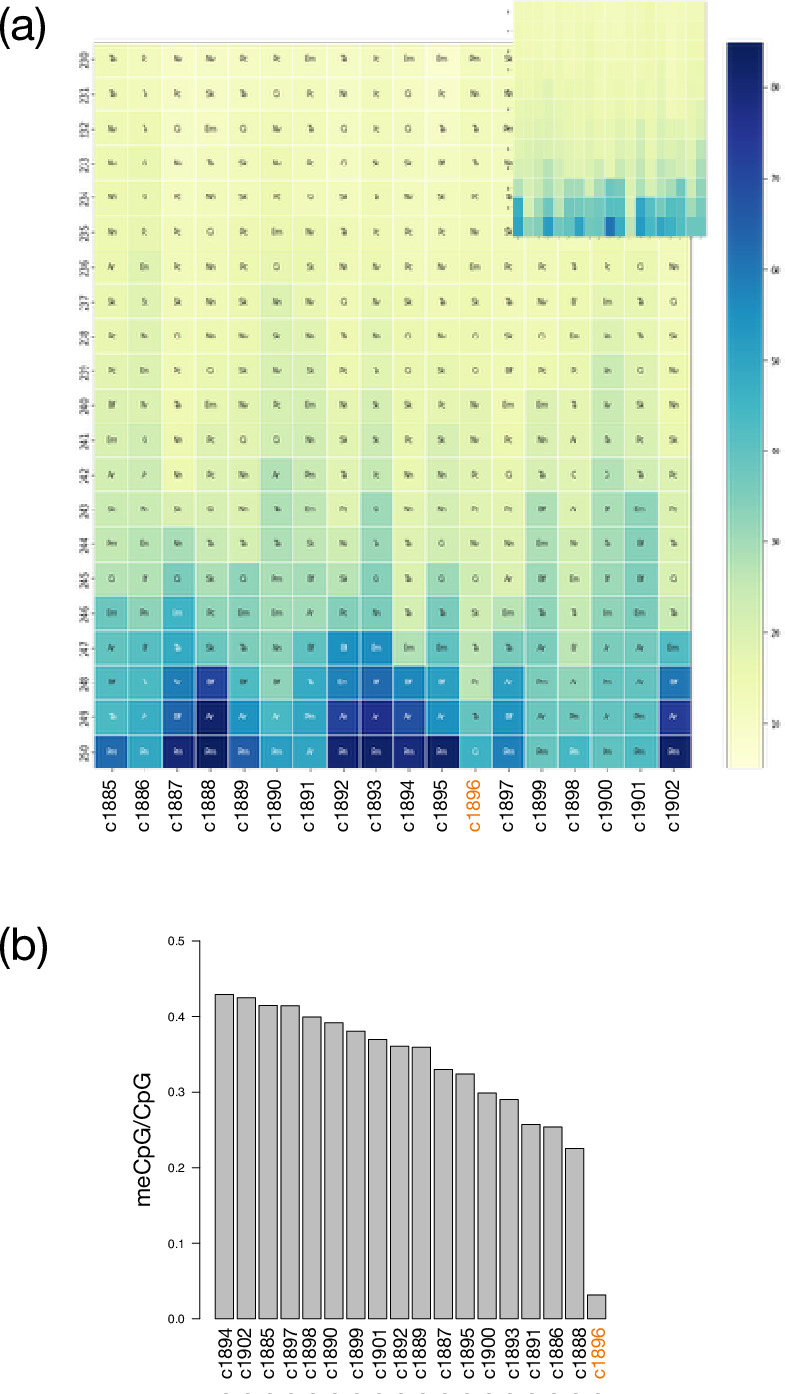
The evolutionary origins of Bilateria remain enigmatic. One of the more enduring proposals highlights similarities between a cnidarian-like planula larva and simple acoel-like flatworms. This idea is based in part on the view of the Xenacoelomorpha as an outgroup to all other bilaterians which are themselves designated the Nephrozoa (protostomes and deuterostomes). Genome data can provide important comparative data and help understand the evolution and biology of enigmatic species better. Here, we assemble and analyze the genome of the simple, marine xenacoelomorph Xenoturbella bocki, a key species for our understanding of early bilaterian evolution. Our highly contiguous genome assembly of X. bocki has a size of ~111 Mbp in 18 chromosome-like scaffolds, with repeat content and intron, exon, and intergenic space comparable to other bilaterian invertebrates. We find X. bocki to have a similar number of genes to other bilaterians and to have retained ancestral metazoan synteny. Key bilaterian signaling pathways are also largely complete and most bilaterian miRNAs are present. Overall, we conclude that X. bocki has a complex genome typical of bilaterians, which does not reflect the apparent simplicity of its body plan that has been so important to proposals that the Xenacoelomorpha are the simple sister group of the rest of the Bilateria.
Keywords: Deuterostomia; Xenacoelomorpha; Xenoturbella; animal evolution; evolutionary biology; genetics; genome analysis; genomics; xenoturbella bocki.
Plain language summary
Xenoturbella bocki is a small marine worm predominantly found on the seafloor of fjords along the west coast of Sweden. This simple organism’s unusual evolutionary history has long intrigued zoologists as it is not clear how it is related to other animal groups. The worm may belong to one of the earliest branches of the animal kingdom, which would explain its simple body. On the other hand, it could be related to a more complex group, the deuterostomes, which includes a wide range of animals, from mammals and birds to sea urchins and starfish. Understanding X. bocki’s evolution could provide valuable insights into how bilaterians evolved as a whole. Unlike its close relatives, the acoelomorphs, X. bocki evolves more slowly, which makes it simpler to study its genome. As a result, it serves as a starting point for investigating the evolutionary processes and genetics underpinning the broader group of bilaterians. To better understand the evolution of X. bocki’s simple body, Schiffer et al. asked whether its genome is simpler or differs in other ways from that of more complex bilaterian organisms. Sequencing the entire X. bocki genome revealed that it has a similar number of genes to that of other animals and includes the genes required for complex biochemical pathways. Reconstructing the worm’s chromosomes – the structures that house genetic information – showed that the X. bocki genes are also distributed in a manner similar to those in other animals. The findings suggest that, despite its simple body plan, X. bocki has a complex genome that is typical of bilaterians. This challenges the idea that X. bocki belongs to a more primitive, simplified sister group to Bilateria and provides a starting point for further studies of how this simple worm evolved.
© 2024, Schiffer et al.
Conflict of interest statement
PS, PN, DL, HR, FL, FM, BF, LB, FS, EH, AZ, PK, KH, SM, MM, HM, RC, RK, PS, MT No competing interests declared
Figures

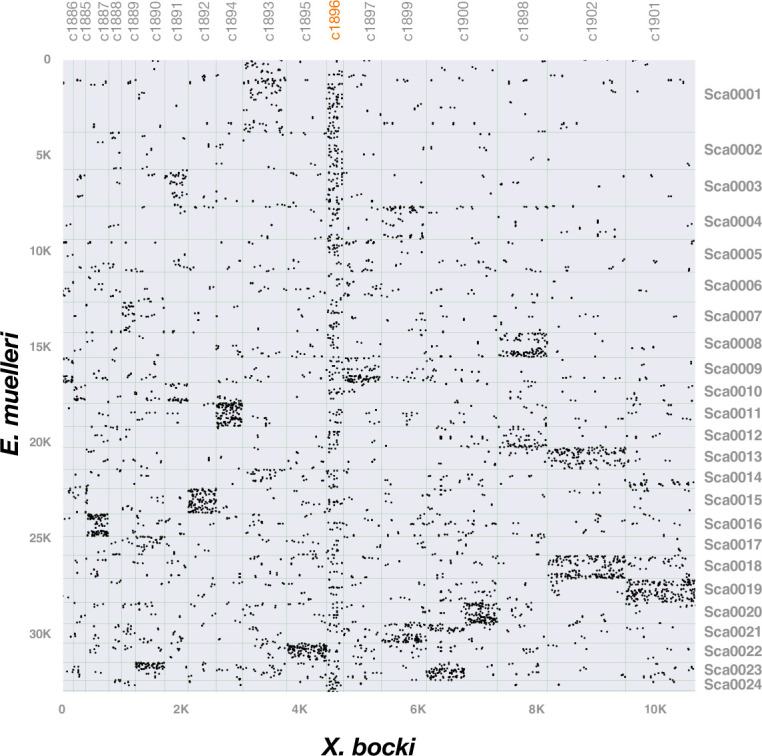
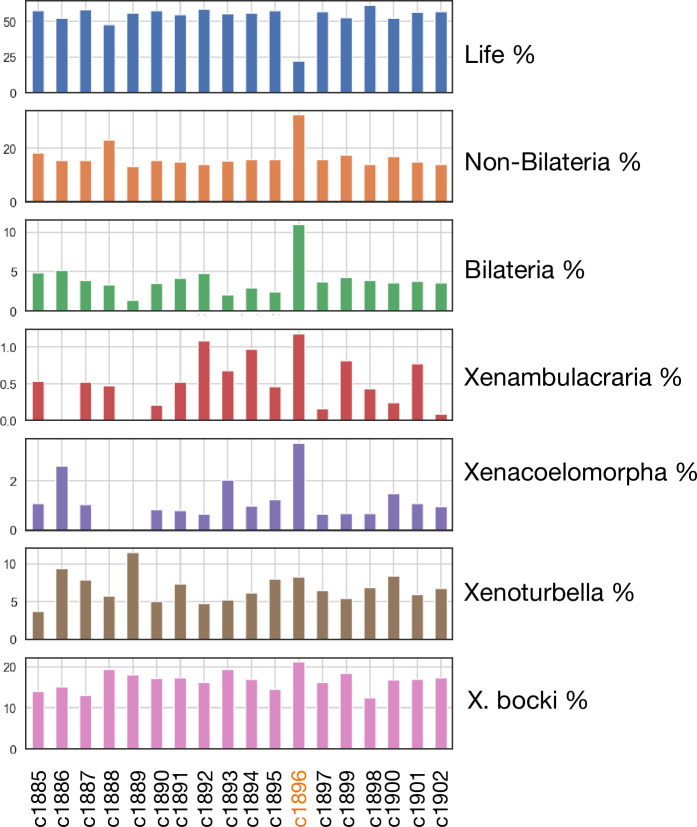
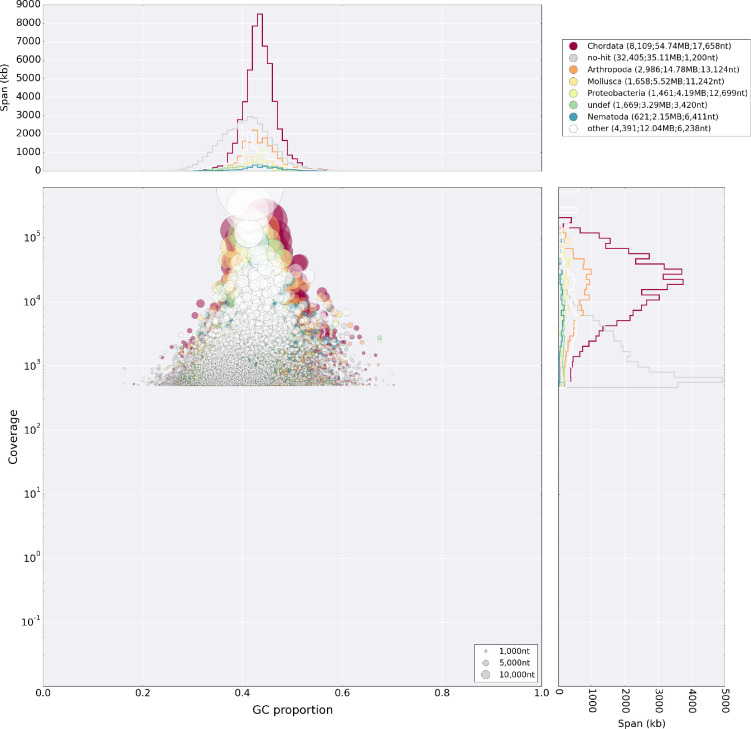
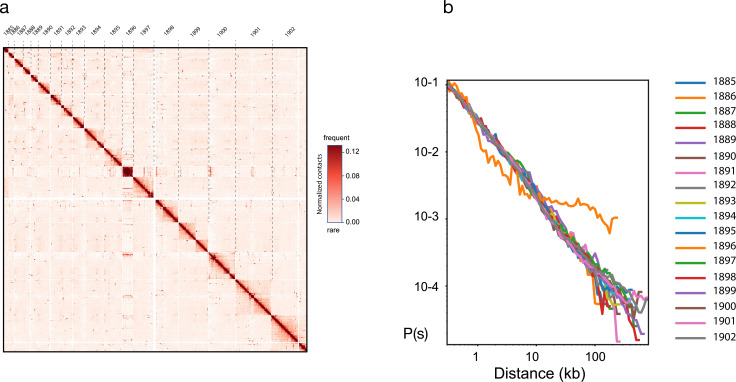
Update of
- doi: 10.1101/2022.06.24.497508
Similar articles
-
Single cell atlas of Xenoturbella bocki highlights limited cell-type complexity.Nat Commun. 2024 Mar 19;15(1):2469. doi: 10.1038/s41467-024-45956-y. Nat Commun. 2024. PMID: 38503762 Free PMC article.
-
A new species of Xenoturbella from the western Pacific Ocean and the evolution of Xenoturbella.BMC Evol Biol. 2017 Dec 18;17(1):245. doi: 10.1186/s12862-017-1080-2. BMC Evol Biol. 2017. PMID: 29249199 Free PMC article.
-
Induced spawning with gamete release from body ruptures during reproduction of Xenoturbella bocki.Commun Biol. 2023 Feb 17;6(1):172. doi: 10.1038/s42003-023-04549-z. Commun Biol. 2023. PMID: 36805023 Free PMC article.
-
What is Xenoturbella?Zoological Lett. 2015 Jul 24;1:22. doi: 10.1186/s40851-015-0018-z. eCollection 2015. Zoological Lett. 2015. PMID: 26605067 Free PMC article. Review.
-
Development of Xenoturbellida.Results Probl Cell Differ. 2019;68:251-258. doi: 10.1007/978-3-030-23459-1_11. Results Probl Cell Differ. 2019. PMID: 31598860 Review.
Cited by
-
The draft genome of the microscopic Nemertoderma westbladi sheds light on the evolution of Acoelomorpha genomes.Front Genet. 2023 Sep 26;14:1244493. doi: 10.3389/fgene.2023.1244493. eCollection 2023. Front Genet. 2023. PMID: 37829276 Free PMC article.
-
Acoelomorph flatworm monophyly is a long-branch attraction artefact obscuring a clade of Acoela and Xenoturbellida.Proc Biol Sci. 2024 Sep;291(2031):20240329. doi: 10.1098/rspb.2024.0329. Epub 2024 Sep 18. Proc Biol Sci. 2024. PMID: 39288803 Free PMC article.
-
Genome assembly of the acoel flatworm Symsagittifera roscoffensis, a model for research on body plan evolution and photosymbiosis.G3 (Bethesda). 2023 Feb 9;13(2):jkac336. doi: 10.1093/g3journal/jkac336. G3 (Bethesda). 2023. PMID: 36542495 Free PMC article.
References
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
-
- Baudry L, Guiglielmoni N, Marie-Nelly H, Cormier A, Marbouty M, Avia K, Mie YL, Godfroy O, Sterck L, Cock JM, Zimmer C, Coelho SM, Koszul R. instaGRAAL: chromosome-level quality scaffolding of genomes using a proximity ligation-based scaffolder. Genome Biology. 2020;21:148. doi: 10.1186/s13059-020-02041-z. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
