

Rescaling protein-protein interactions improves Martini 3 for flexible proteins in solution
- PMID: 39103332
- PMCID: PMC11300910
- DOI: 10.1038/s41467-024-50647-9
Rescaling protein-protein interactions improves Martini 3 for flexible proteins in solution
Abstract
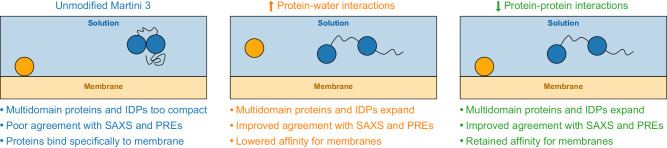
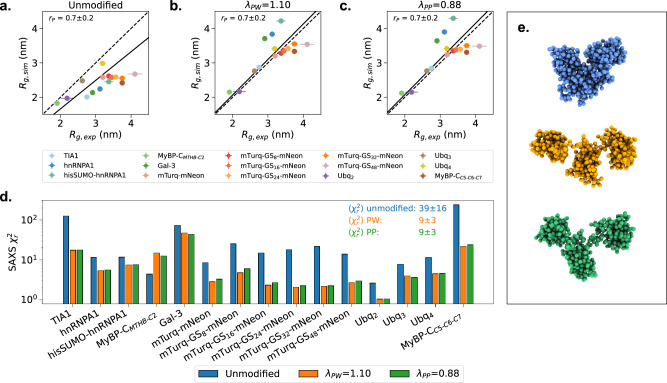
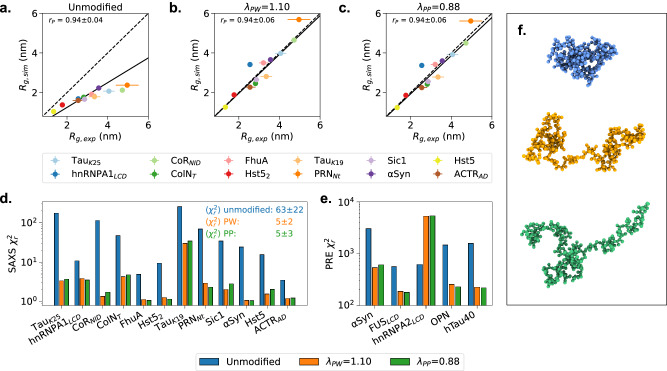
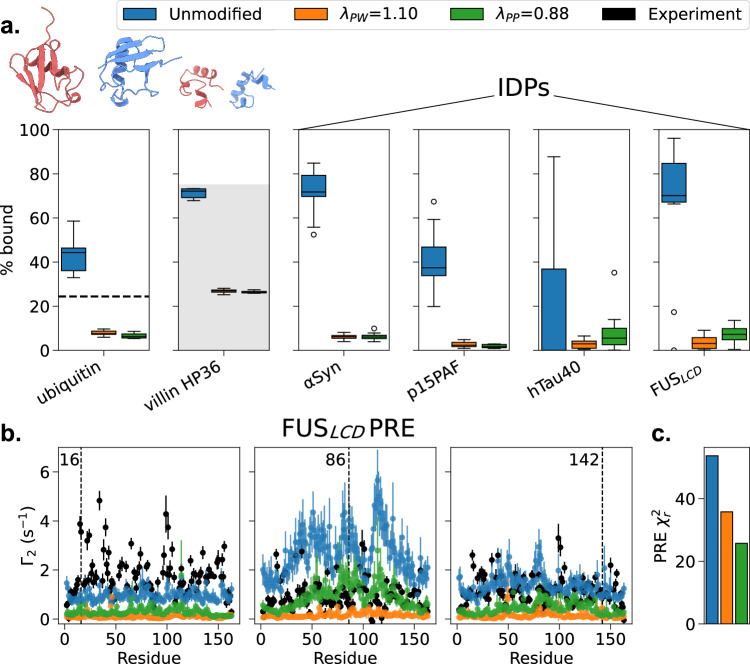
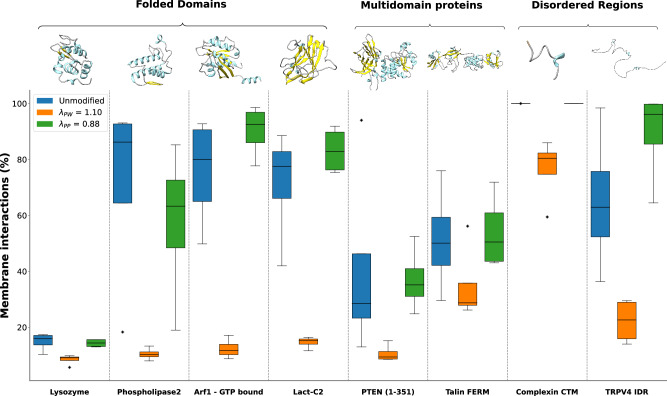
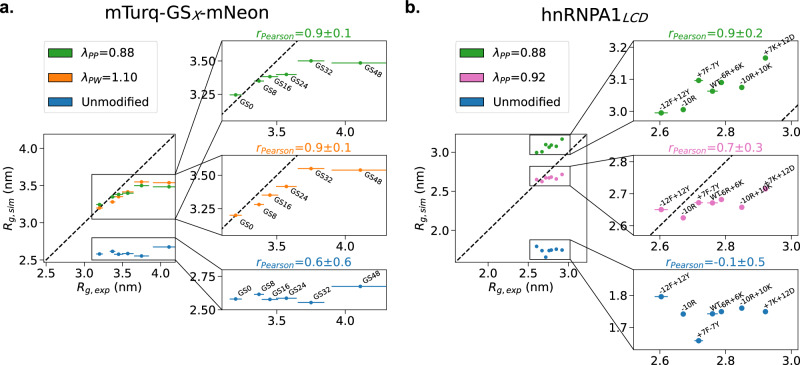
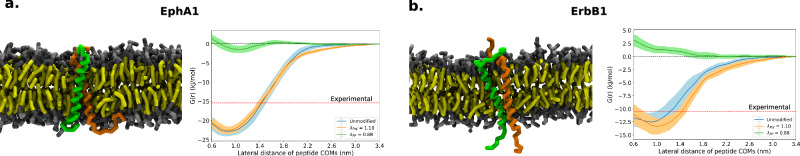
Multidomain proteins with flexible linkers and disordered regions play important roles in many cellular processes, but characterizing their conformational ensembles is difficult. We have previously shown that the coarse-grained model, Martini 3, produces too compact ensembles in solution, that may in part be remedied by strengthening protein-water interactions. Here, we show that decreasing the strength of protein-protein interactions leads to improved agreement with experimental data on a wide set of systems. We show that the 'symmetry' between rescaling protein-water and protein-protein interactions breaks down when studying interactions with or within membranes; rescaling protein-protein interactions better preserves the binding specificity of proteins with lipid membranes, whereas rescaling protein-water interactions preserves oligomerization of transmembrane helices. We conclude that decreasing the strength of protein-protein interactions improves the accuracy of Martini 3 for IDPs and multidomain proteins, both in solution and in the presence of a lipid membrane.
© 2024. The Author(s).
Conflict of interest statement
K.L.-L. holds stock options in and is a consultant for Peptone Ltd. All other authors declare no competing interests.
Figures
Similar articles
-
Improving Martini 3 for Disordered and Multidomain Proteins.J Chem Theory Comput. 2022 Apr 12;18(4):2033-2041. doi: 10.1021/acs.jctc.1c01042. Epub 2022 Apr 4. J Chem Theory Comput. 2022. PMID: 35377637
-
Curvature Footprints of Transmembrane Proteins in Simulations with the Martini Force Field.J Phys Chem B. 2024 Jun 27;128(25):5987-5994. doi: 10.1021/acs.jpcb.4c01385. Epub 2024 Jun 11. J Phys Chem B. 2024. PMID: 38860934
-
Recalibration of MARTINI-3 Parameters for Improved Interactions between Peripheral Proteins and Lipid Bilayers.J Chem Theory Comput. 2024 Nov 12;20(21):9673-9686. doi: 10.1021/acs.jctc.4c00645. Epub 2024 Nov 3. J Chem Theory Comput. 2024. PMID: 39491480
-
Assessing the Martini 3 protein model: A review of its path and potential.Biochim Biophys Acta Proteins Proteom. 2024 Jul 1;1872(4):141014. doi: 10.1016/j.bbapap.2024.141014. Epub 2024 Apr 25. Biochim Biophys Acta Proteins Proteom. 2024. PMID: 38670324 Review.
-
An overview of molecular dynamics simulations of oxidized lipid systems, with a comparison of ELBA and MARTINI force fields for coarse grained lipid simulations.Biochim Biophys Acta. 2016 Oct;1858(10):2498-2511. doi: 10.1016/j.bbamem.2016.03.031. Epub 2016 Apr 6. Biochim Biophys Acta. 2016. PMID: 27058982 Review.
Cited by
-
Sequence and structural determinants of RNAPII CTD phase-separation and phosphorylation by CDK7.Nat Commun. 2024 Oct 24;15(1):9163. doi: 10.1038/s41467-024-53305-2. Nat Commun. 2024. PMID: 39448580 Free PMC article.
-
A Coarse-Grained SPICA Makeover for Solvated and Bare Sodium and Chloride Ions.J Chem Theory Comput. 2024 Sep 10;20(17):7624-7634. doi: 10.1021/acs.jctc.4c00529. Epub 2024 Aug 19. J Chem Theory Comput. 2024. PMID: 39160094 Free PMC article.
-
Coarse-Grained Simulations of Adeno-Associated Virus and Its Receptor Reveal Influences on Membrane Lipid Organization and Curvature.J Phys Chem B. 2024 Oct 17;128(41):10139-10153. doi: 10.1021/acs.jpcb.4c03087. Epub 2024 Oct 2. J Phys Chem B. 2024. PMID: 39356546 Free PMC article.
-
A coarse-grained model for disordered and multi-domain proteins.Protein Sci. 2024 Nov;33(11):e5172. doi: 10.1002/pro.5172. Protein Sci. 2024. PMID: 39412378 Free PMC article.
References
-
- Thomasen, F. E. & Larsen, K. L. Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. Biochem. Soc. Trans.50, 541–554 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
