

The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases
- PMID: 39000044
- PMCID: PMC11241570
- DOI: 10.3390/ijms25136936
The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases
Abstract
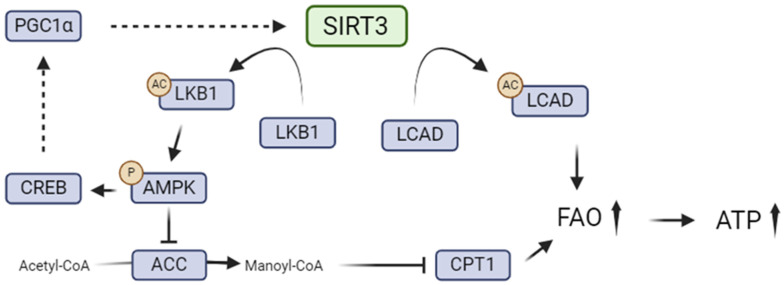
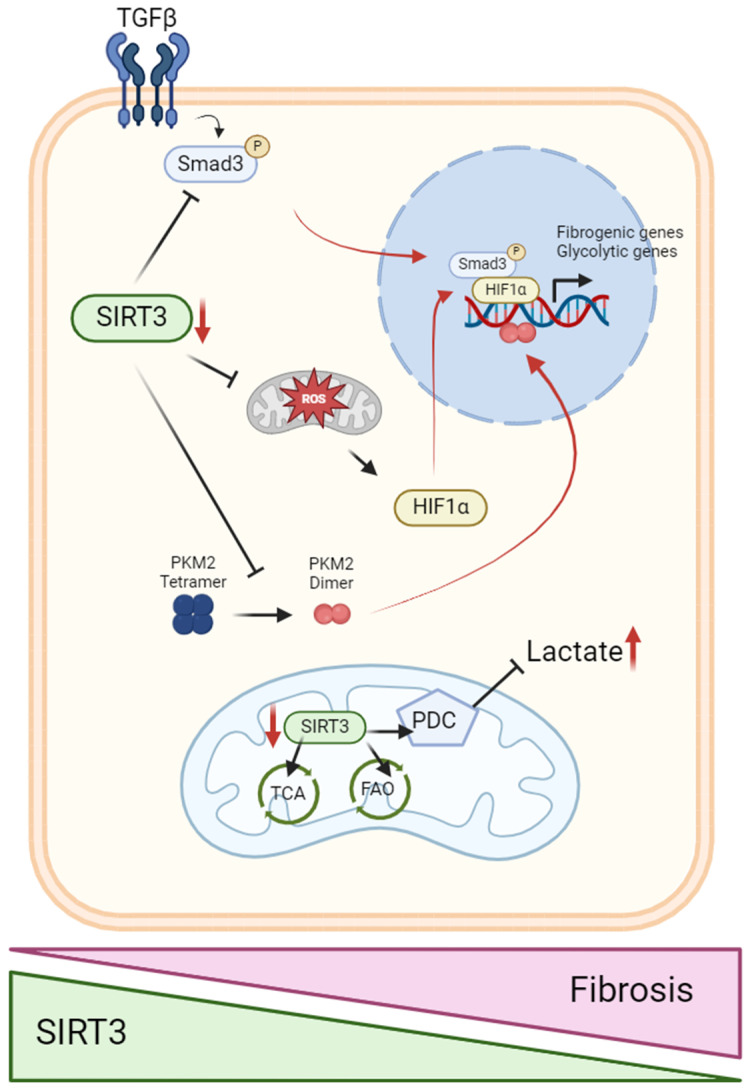
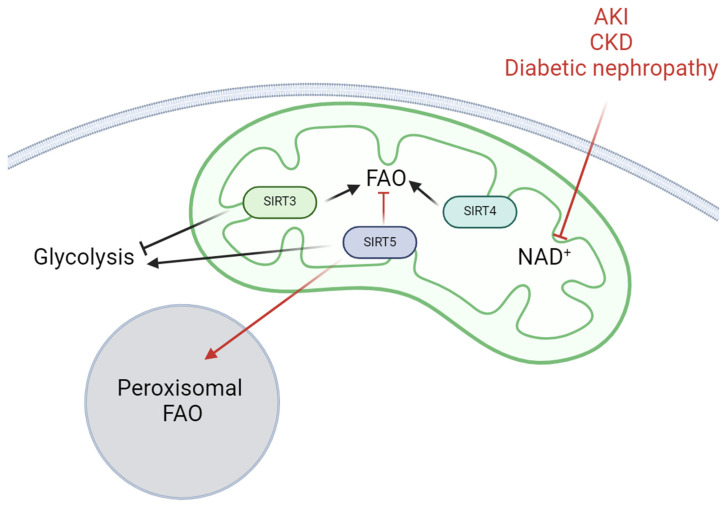
Kidney diseases, including chronic kidney disease (CKD), diabetic nephropathy, and acute kidney injury (AKI), represent a significant global health burden. The kidneys are metabolically very active organs demanding a large amount of ATP. They are composed of highly specialized cell types in the glomerulus and subsequent tubular compartments which fine-tune metabolism to meet their numerous and diverse functions. Defective renal cell metabolism, including altered fatty acid oxidation or glycolysis, has been linked to both AKI and CKD. Mitochondria play a vital role in renal metabolism, and emerging research has identified mitochondrial sirtuins (SIRT3, SIRT4 and SIRT5) as key regulators of renal cell metabolic adaptation, especially SIRT3. Sirtuins belong to an evolutionarily conserved family of mainly NAD+-dependent deacetylases, deacylases, and ADP-ribosyl transferases. Their dependence on NAD+, used as a co-substrate, directly links their enzymatic activity to the metabolic status of the cell. In the kidney, SIRT3 has been described to play crucial roles in the regulation of mitochondrial function, and the antioxidative and antifibrotic response. SIRT3 has been found to be constantly downregulated in renal diseases. Genetic or pharmacologic upregulation of SIRT3 has also been associated with beneficial renal outcomes. Importantly, experimental pieces of evidence suggest that SIRT3 may act as an important energy sensor in renal cells by regulating the activity of key enzymes involved in metabolic adaptation. Activation of SIRT3 may thus represent an interesting strategy to ameliorate renal cell energetics. In this review, we discuss the roles of SIRT3 in lipid and glucose metabolism and in mediating a metabolic switch in a physiological and pathological context. Moreover, we highlight the emerging significance of other mitochondrial sirtuins, SIRT4 and SIRT5, in renal metabolism. Understanding the role of mitochondrial sirtuins in kidney diseases may also open new avenues for innovative and efficient therapeutic interventions and ultimately improve the management of renal injuries.
Keywords: SIRT3; SIRT4; SIRT5; glucose metabolism; kidney disease; lipid metabolism; lipotoxicity; metabolic switch; mitochondrial homeostasis; sirtuins.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Overexpression of mitochondrial sirtuins alters glycolysis and mitochondrial function in HEK293 cells.PLoS One. 2014 Aug 28;9(8):e106028. doi: 10.1371/journal.pone.0106028. eCollection 2014. PLoS One. 2014. PMID: 25165814 Free PMC article.
-
Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5.J Mol Biol. 2008 Oct 10;382(3):790-801. doi: 10.1016/j.jmb.2008.07.048. Epub 2008 Jul 25. J Mol Biol. 2008. PMID: 18680753
-
Mitochondrial sirtuins: Energy dynamics and cancer metabolism.Mol Cells. 2024 Feb;47(2):100029. doi: 10.1016/j.mocell.2024.100029. Epub 2024 Feb 6. Mol Cells. 2024. PMID: 38331199 Free PMC article. Review.
-
Mitochondrial sirtuins in the rat adrenal gland: location within the glands of males and females, hormonal and developmental regulation of gene expressions.Folia Histochem Cytobiol. 2017;55(4):190-202. doi: 10.5603/FHC.a2017.0020. Epub 2017 Dec 20. Folia Histochem Cytobiol. 2017. PMID: 29261224
-
The role of mitochondrial sirtuins in health and disease.Free Radic Biol Med. 2016 Nov;100:164-174. doi: 10.1016/j.freeradbiomed.2016.04.197. Epub 2016 May 6. Free Radic Biol Med. 2016. PMID: 27164052 Review.
Cited by
-
Redox Imbalance and Mitochondrial Abnormalities in Kidney Disease-Volume II.Biomolecules. 2024 Aug 9;14(8):973. doi: 10.3390/biom14080973. Biomolecules. 2024. PMID: 39199361 Free PMC article.
-
Activation of sirtuin 3 and maintenance of mitochondrial homeostasis by artemisinin protect against diclofenac-induced kidney injury in rats.Naunyn Schmiedebergs Arch Pharmacol. 2024 Nov 23. doi: 10.1007/s00210-024-03620-8. Online ahead of print. Naunyn Schmiedebergs Arch Pharmacol. 2024. PMID: 39579210
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
