

Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation
- PMID: 38992677
- PMCID: PMC11241886
- DOI: 10.1186/s13040-024-00374-0
Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation
Abstract
Background: Changing cell-type proportions can confound studies of differential gene expression or DNA methylation (DNAm) from peripheral blood mononuclear cells (PBMCs). We examined how cell-type proportions derived from the transcriptome versus the methylome (DNAm) influence estimates of differentially expressed genes (DEGs) and differentially methylated positions (DMPs).
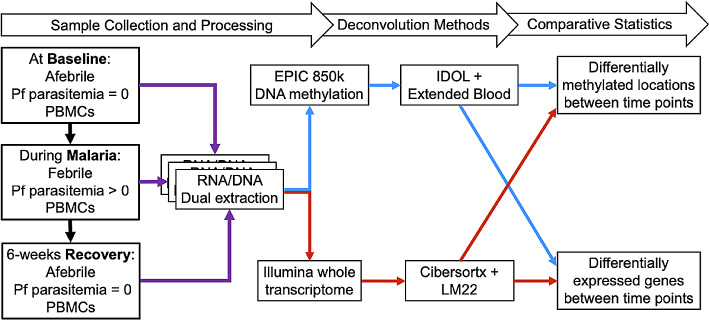
Methods: Transcriptome and DNAm data were obtained from PBMC RNA and DNA of Kenyan children (n = 8) before, during, and 6 weeks following uncomplicated malaria. DEGs and DMPs between time points were detected using cell-type adjusted modeling with Cibersortx or IDOL, respectively.
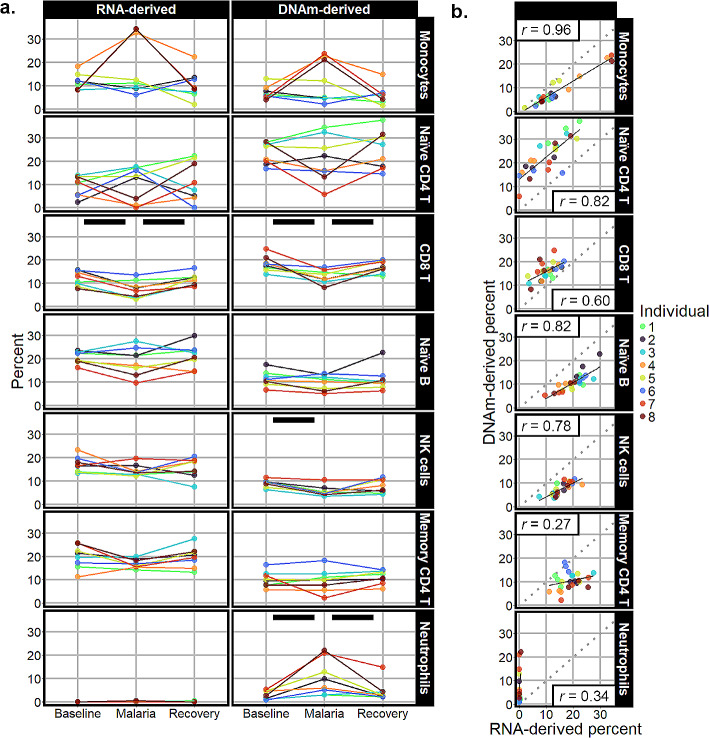
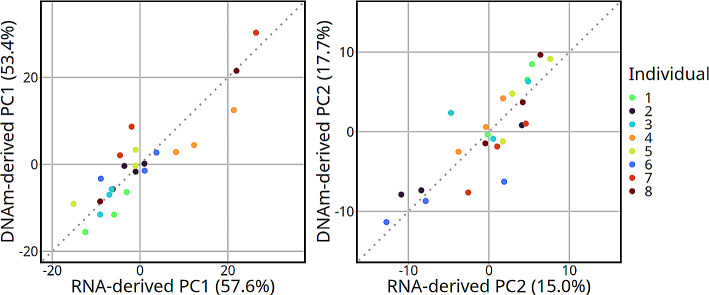
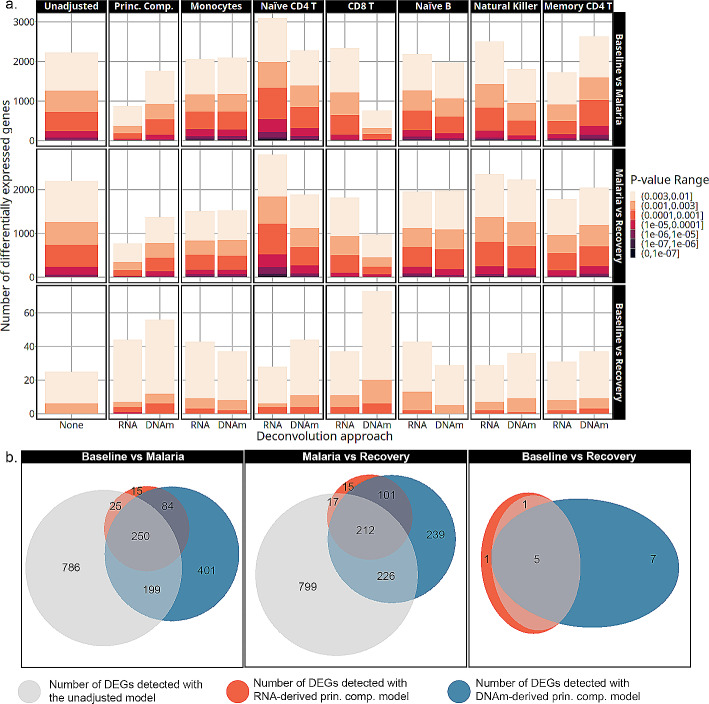
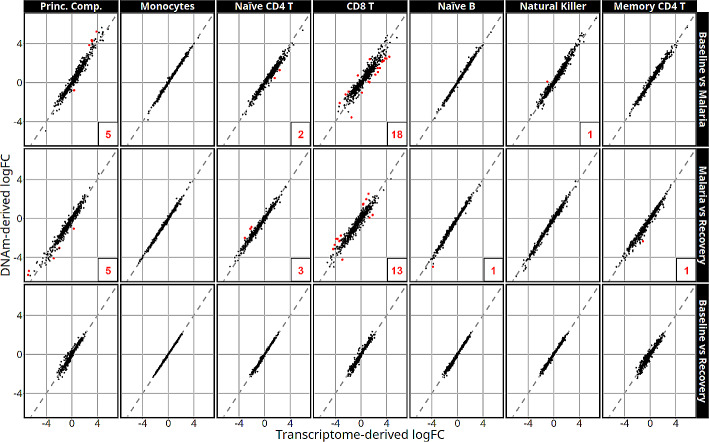
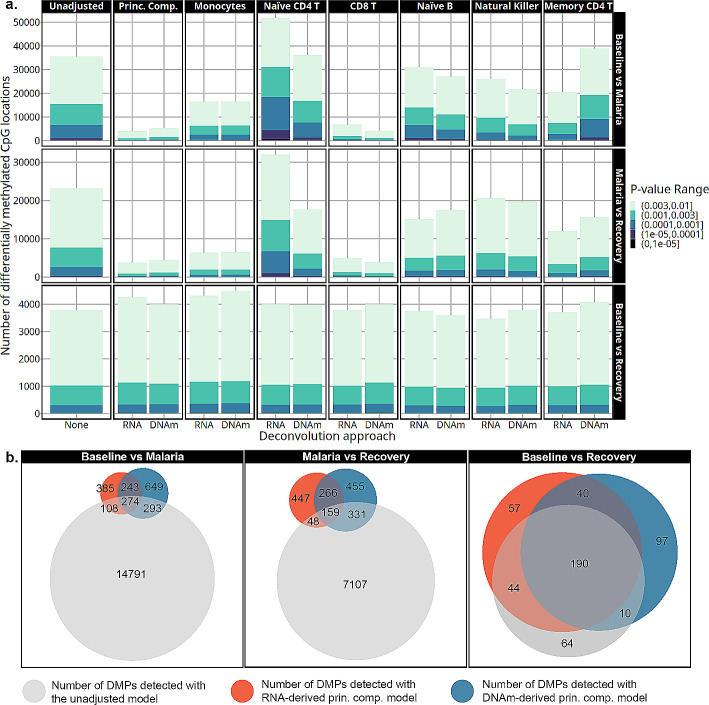
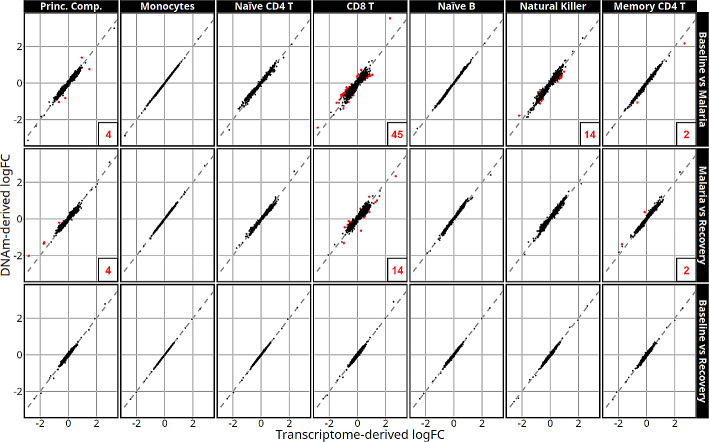
Results: Most major cell types and principal components had moderate to high correlation between the two deconvolution methods (r = 0.60-0.96). Estimates of cell-type proportions and DEGs or DMPs were largely unaffected by the method, with the greatest discrepancy in the estimation of neutrophils.
Conclusion: Variation in cell-type proportions is captured similarly by both transcriptomic and methylome deconvolution methods for most major cell types.
Keywords: Deconvolution; Gene expression; PBMC.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation.Res Sq [Preprint]. 2024 Apr 3:rs.3.rs-3992113. doi: 10.21203/rs.3.rs-3992113/v1. Res Sq. 2024. Update in: BioData Min. 2024 Jul 11;17(1):21. doi: 10.1186/s13040-024-00374-0. PMID: 38645047 Free PMC article. Updated. Preprint.
Similar articles
-
Transcriptome- and DNA methylation-based cell-type deconvolutions produce similar estimates of differential gene expression and differential methylation.Res Sq [Preprint]. 2024 Apr 3:rs.3.rs-3992113. doi: 10.21203/rs.3.rs-3992113/v1. Res Sq. 2024. Update in: BioData Min. 2024 Jul 11;17(1):21. doi: 10.1186/s13040-024-00374-0. PMID: 38645047 Free PMC article. Updated. Preprint.
-
Exploration of the pathogenesis of Sjögren's syndrome via DNA methylation and transcriptome analyses.Clin Rheumatol. 2022 Sep;41(9):2765-2777. doi: 10.1007/s10067-022-06200-4. Epub 2022 May 13. Clin Rheumatol. 2022. PMID: 35562622
-
An assessment of compositional methods for the analysis of DNA methylation-based deconvolution estimates.Epigenomics. 2024;16(15-16):1067-1080. doi: 10.1080/17501911.2024.2379242. Epub 2024 Aug 2. Epigenomics. 2024. PMID: 39093129
-
Exploration of the sputum methylome and omics deconvolution by quadratic programming in molecular profiling of asthma and COPD: the road to sputum omics 2.0.Respir Res. 2020 Oct 19;21(1):274. doi: 10.1186/s12931-020-01544-4. Respir Res. 2020. PMID: 33076907 Free PMC article.
-
Cell-type deconvolution from DNA methylation: a review of recent applications.Hum Mol Genet. 2017 Oct 1;26(R2):R216-R224. doi: 10.1093/hmg/ddx275. Hum Mol Genet. 2017. PMID: 28977446 Free PMC article. Review.
Cited by
-
Transcriptomic and Proteomic Insights into Host Immune Responses in Pediatric Severe Malarial Anemia: Dysregulation in HSP60-70-TLR2/4 Signaling and Altered Glutamine Metabolism.Pathogens. 2024 Oct 3;13(10):867. doi: 10.3390/pathogens13100867. Pathogens. 2024. PMID: 39452740 Free PMC article.
-
Inflammatory breast cancer microenvironment repertoire based on DNA methylation data deconvolution reveals actionable targets to enhance the treatment efficacy.J Transl Med. 2024 Aug 5;22(1):735. doi: 10.1186/s12967-024-05553-5. J Transl Med. 2024. PMID: 39103878 Free PMC article.
References
-
- Moncunill G, Scholzen A, Mpina M, Nhabomba A, Hounkpatin AB, Osaba L et al. Antigen-stimulated PBMC transcriptional protective signatures for malaria immunization. Sci Transl Med. 2020;12(543). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
