

Cleavage site-directed antibodies reveal the prion protein in humans is shed by ADAM10 at Y226 and associates with misfolded protein deposits in neurodegenerative diseases
- PMID: 38980441
- PMCID: PMC11233397
- DOI: 10.1007/s00401-024-02763-5
Cleavage site-directed antibodies reveal the prion protein in humans is shed by ADAM10 at Y226 and associates with misfolded protein deposits in neurodegenerative diseases
Abstract
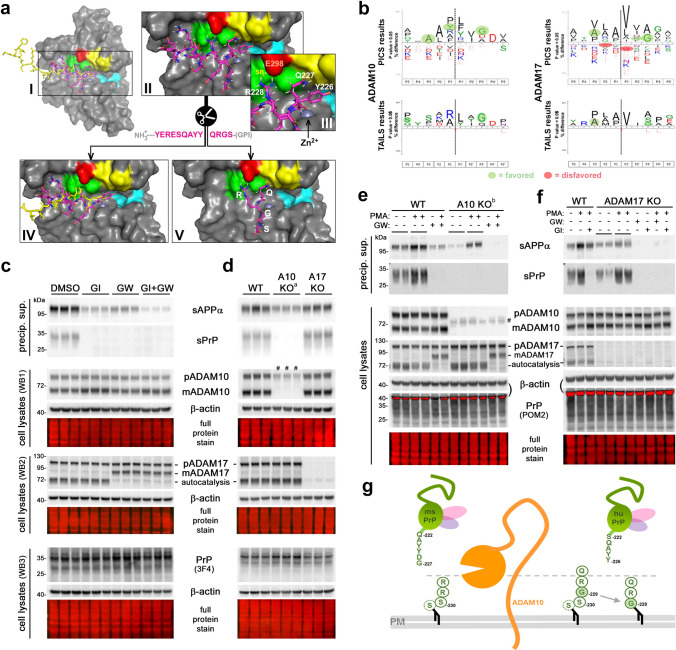
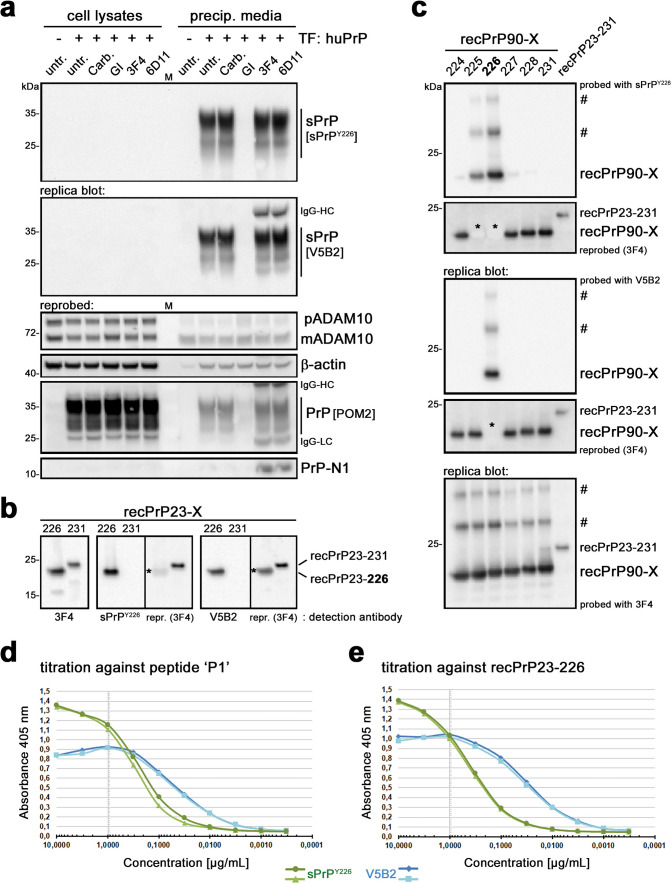
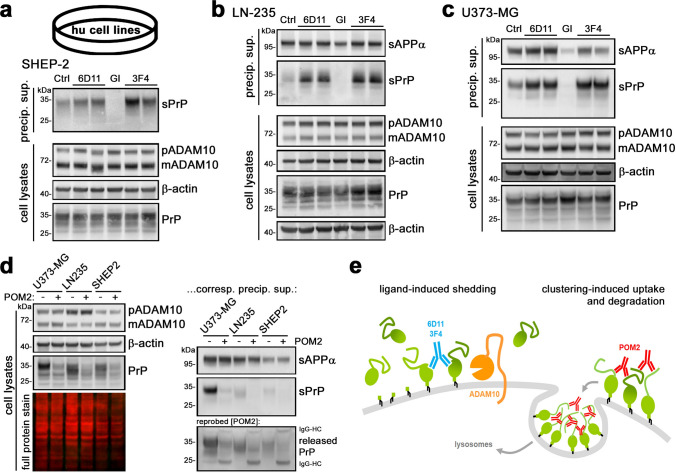
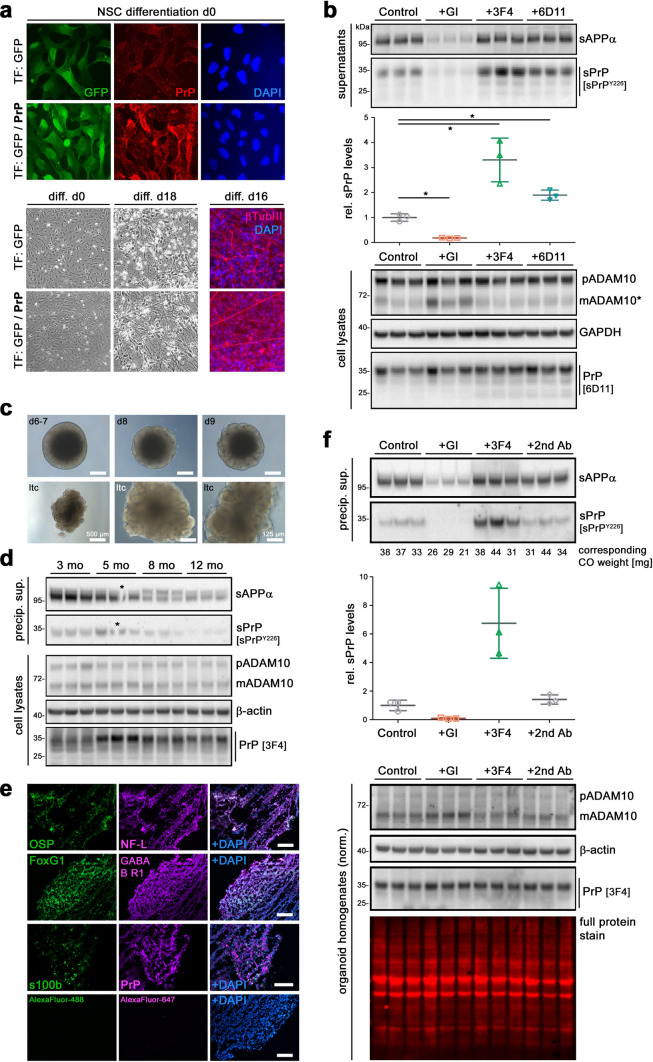
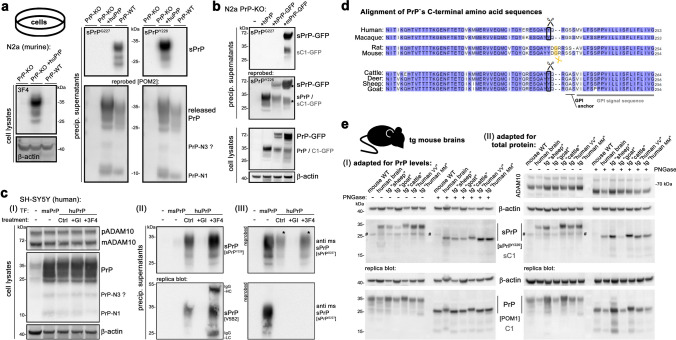
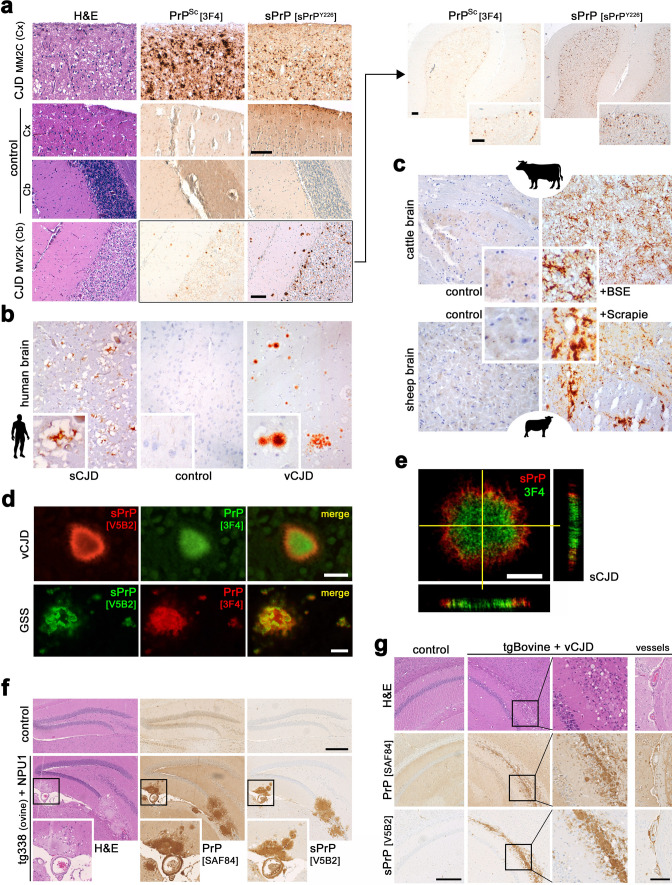
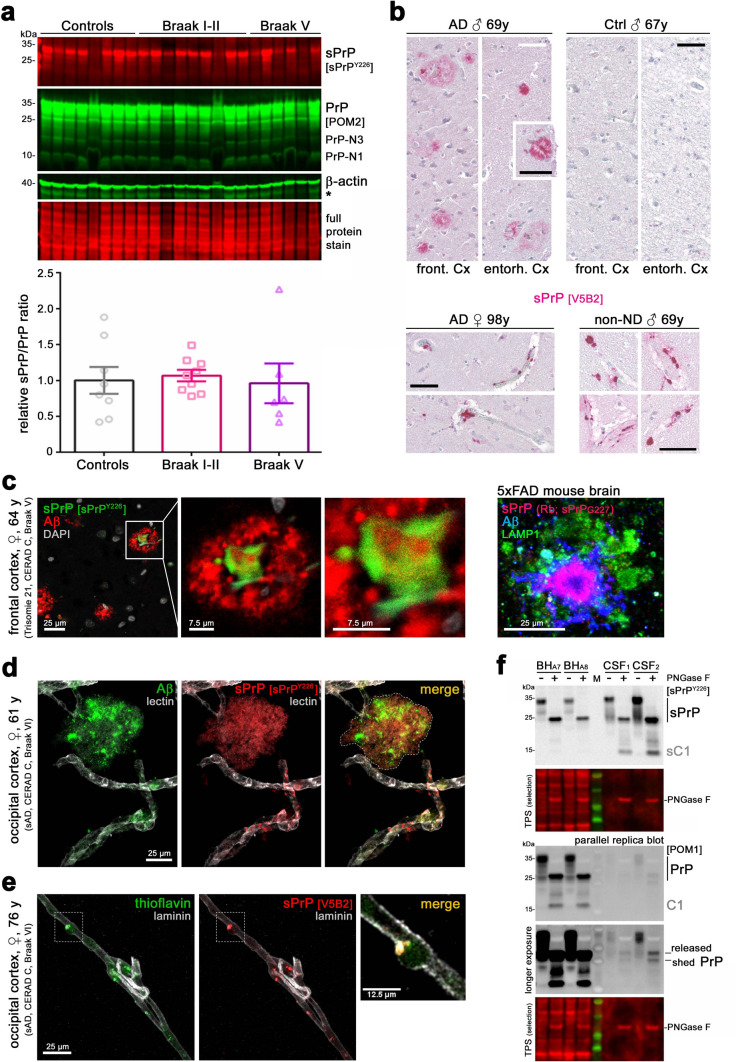
Proteolytic cell surface release ('shedding') of the prion protein (PrP), a broadly expressed GPI-anchored glycoprotein, by the metalloprotease ADAM10 impacts on neurodegenerative and other diseases in animal and in vitro models. Recent studies employing the latter also suggest shed PrP (sPrP) to be a ligand in intercellular communication and critically involved in PrP-associated physiological tasks. Although expectedly an evolutionary conserved event, and while soluble forms of PrP are present in human tissues and body fluids, for the human body neither proteolytic PrP shedding and its cleavage site nor involvement of ADAM10 or the biological relevance of this process have been demonstrated thus far. In this study, cleavage site prediction and generation (plus detailed characterization) of sPrP-specific antibodies enabled us to identify PrP cleaved at tyrosin 226 as the physiological and apparently strictly ADAM10-dependent shed form in humans. Using cell lines, neural stem cells and brain organoids, we show that shedding of human PrP can be stimulated by PrP-binding ligands without targeting the protease, which may open novel therapeutic perspectives. Site-specific antibodies directed against human sPrP also detect the shed form in brains of cattle, sheep and deer, hence in all most relevant species naturally affected by fatal and transmissible prion diseases. In human and animal prion diseases, but also in patients with Alzheimer`s disease, sPrP relocalizes from a physiological diffuse tissue pattern to intimately associate with extracellular aggregated deposits of misfolded proteins characteristic for the respective pathological condition. Findings and research tools presented here will accelerate novel insight into the roles of PrP shedding (as a process) and sPrP (as a released factor) in neurodegeneration and beyond.
Keywords: Alzheimer’s disease; Dementia; Extracellular vesicles; Neuroprotection; Prions; Proteolytic processing.
© 2024. The Author(s).
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures

Similar articles
-
Anchorless risk or released benefit? An updated view on the ADAM10-mediated shedding of the prion protein.Cell Tissue Res. 2023 Apr;392(1):215-234. doi: 10.1007/s00441-022-03582-4. Epub 2022 Jan 27. Cell Tissue Res. 2023. PMID: 35084572 Free PMC article. Review.
-
Inducing prion protein shedding as a neuroprotective and regenerative approach in pathological conditions of the brain: from theory to facts.Neural Regen Res. 2023 Sep;18(9):1869-1875. doi: 10.4103/1673-5374.366496. Neural Regen Res. 2023. PMID: 36926701 Free PMC article. Review.
-
Proteolytic shedding of the prion protein via activation of metallopeptidase ADAM10 reduces cellular binding and toxicity of amyloid-β oligomers.J Biol Chem. 2019 Apr 26;294(17):7085-7097. doi: 10.1074/jbc.RA118.005364. Epub 2019 Mar 14. J Biol Chem. 2019. PMID: 30872401 Free PMC article.
-
Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein.Mol Neurodegener. 2018 Apr 6;13(1):18. doi: 10.1186/s13024-018-0248-6. Mol Neurodegener. 2018. PMID: 29625583 Free PMC article.
-
The sheddase ADAM10 is a potent modulator of prion disease.Elife. 2015 Feb 5;4:e04260. doi: 10.7554/eLife.04260. Elife. 2015. PMID: 25654651 Free PMC article.
References
-
- Aguilar-Calvo P, Sevillano AM, Bapat J, Soldau K, Sandoval DR, Altmeppen HC, et al. Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol. 2020;139:527–546. doi: 10.1007/s00401-019-02085-x. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- p19050/Alzheimer Forschung Initiative
- CRC877 "Proteolysis as a regulatory event in pathophysiology" project A12/Deutsche Forschungsgemeinschaft
- Munich Cluster for Systems Neurology (EXC 2145 SyNergy/Deutsche Forschungsgemeinschaft
- ID 390857198)/Deutsche Forschungsgemeinschaft
- TA 167/6-3/Deutsche Forschungsgemeinschaft
- EXC 2033 - 390677874 - RESOLV/Deutsche Forschungsgemeinschaft
- SCHA 2248/2-1/Deutsche Forschungsgemeinschaft
- grant #202108080249/China Scholarship Council
- grant number: L3-3435/Slovene Research Agency
- grant number: L3-6006/Slovene Research Agency
- grant number: L3-0206/Slovene Research Agency
- grant number: P4-0176/Slovene Research Agency
- grant number: P3-0171/Slovene Research Agency
- PhD student funding/Slovene Research Agency
- Marie Skłodowska Curie grant #101030402/Horizon 2020 Framework Programme
LinkOut - more resources
Full Text Sources
Medical
Research Materials
