

Noncanonical Amino Acids in Biocatalysis
- PMID: 38959423
- PMCID: PMC11273360
- DOI: 10.1021/acs.chemrev.4c00120
Noncanonical Amino Acids in Biocatalysis
Abstract
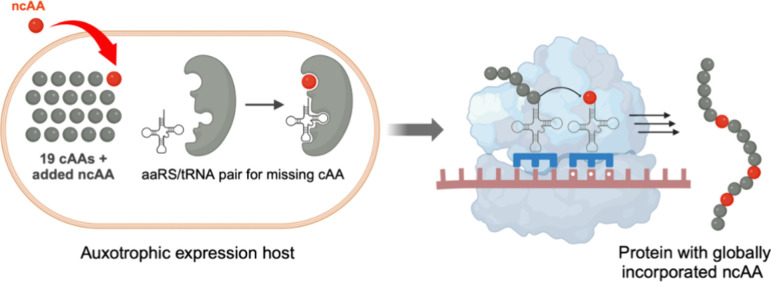
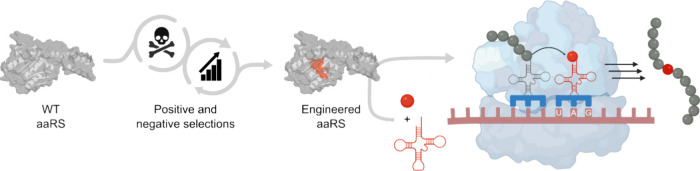
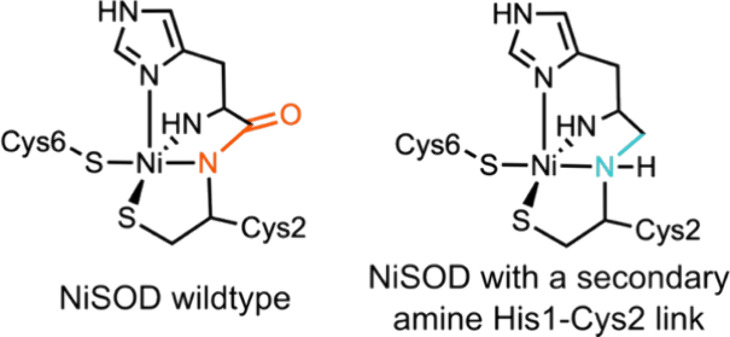
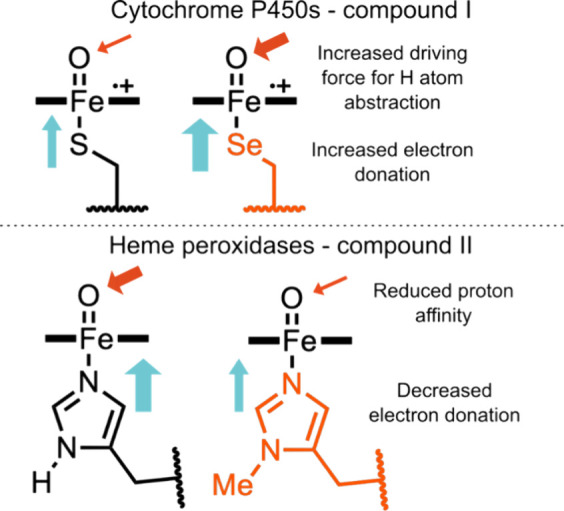
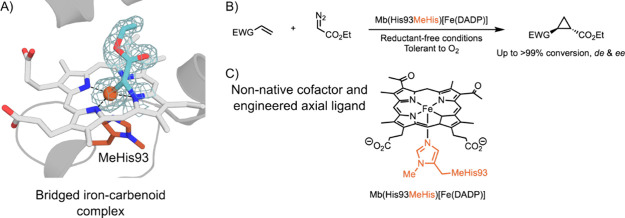
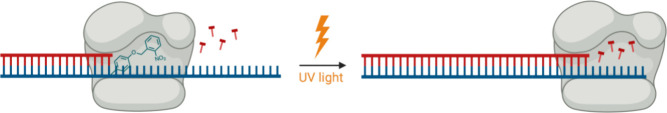
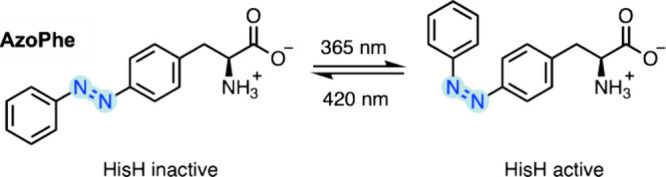
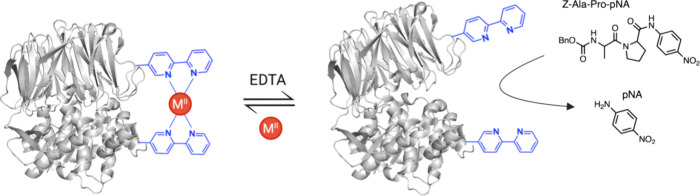
In recent years, powerful genetic code reprogramming methods have emerged that allow new functional components to be embedded into proteins as noncanonical amino acid (ncAA) side chains. In this review, we will illustrate how the availability of an expanded set of amino acid building blocks has opened a wealth of new opportunities in enzymology and biocatalysis research. Genetic code reprogramming has provided new insights into enzyme mechanisms by allowing introduction of new spectroscopic probes and the targeted replacement of individual atoms or functional groups. NcAAs have also been used to develop engineered biocatalysts with improved activity, selectivity, and stability, as well as enzymes with artificial regulatory elements that are responsive to external stimuli. Perhaps most ambitiously, the combination of genetic code reprogramming and laboratory evolution has given rise to new classes of enzymes that use ncAAs as key catalytic elements. With the framework for developing ncAA-containing biocatalysts now firmly established, we are optimistic that genetic code reprogramming will become a progressively more powerful tool in the armory of enzyme designers and engineers in the coming years.
Conflict of interest statement
The authors declare no competing financial interest.
Figures












Similar articles
-
Enzymes with noncanonical amino acids.Curr Opin Chem Biol. 2020 Apr;55:136-144. doi: 10.1016/j.cbpa.2020.01.006. Epub 2020 Mar 9. Curr Opin Chem Biol. 2020. PMID: 32163871 Review.
-
Engineering enzyme activity using an expanded amino acid alphabet.Protein Eng Des Sel. 2023 Jan 21;36:gzac013. doi: 10.1093/protein/gzac013. Protein Eng Des Sel. 2023. PMID: 36370045 Free PMC article.
-
Engineering enzymes for noncanonical amino acid synthesis.Chem Soc Rev. 2018 Dec 21;47(24):8980-8997. doi: 10.1039/c8cs00665b. Epub 2018 Oct 3. Chem Soc Rev. 2018. PMID: 30280154 Free PMC article. Review.
-
Noncanonical Amino Acids: Bringing New-to-Nature Functionalities to Biocatalysis.Chem Rev. 2024 Oct 9;124(19):10877-10923. doi: 10.1021/acs.chemrev.4c00136. Epub 2024 Sep 27. Chem Rev. 2024. PMID: 39329413 Free PMC article. Review.
-
Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology.Angew Chem Int Ed Engl. 2017 Aug 7;56(33):9680-9703. doi: 10.1002/anie.201610129. Epub 2017 Jul 17. Angew Chem Int Ed Engl. 2017. PMID: 28085996 Review.
References
-
- Punekar N. S.ENZYMES: Catalysis, Kinetics and Mechanisms; Springer Nature Singapore, 2018.10.1007/978-981-13-0785-0 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
