

Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
- PMID: 38932250
- PMCID: PMC11209099
- DOI: 10.3390/v16060958
Epidemiological and Genetic Characteristics of Respiratory Viral Coinfections with Different Variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
Abstract
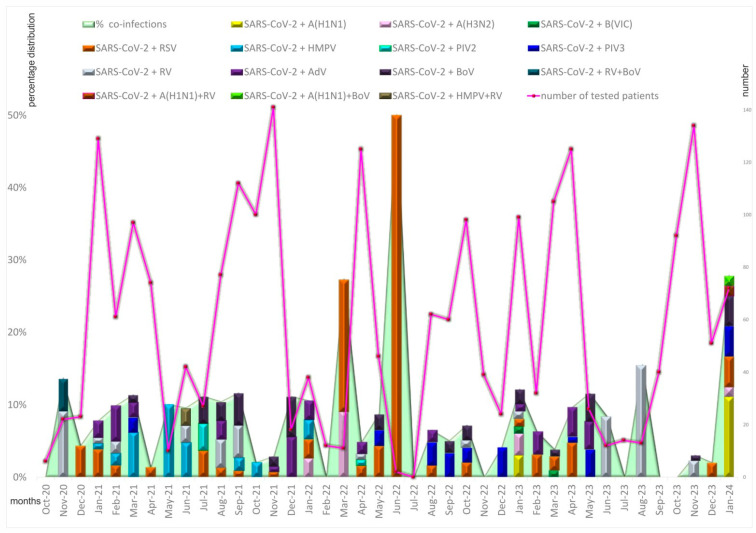
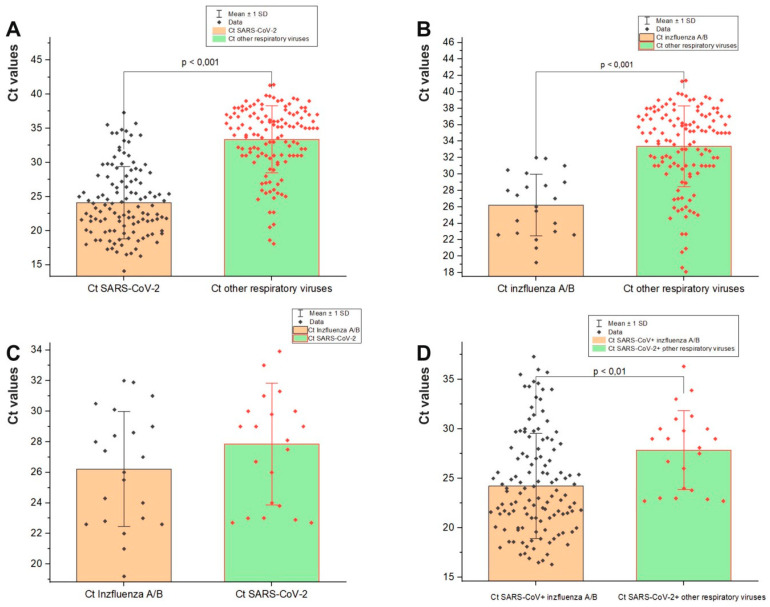
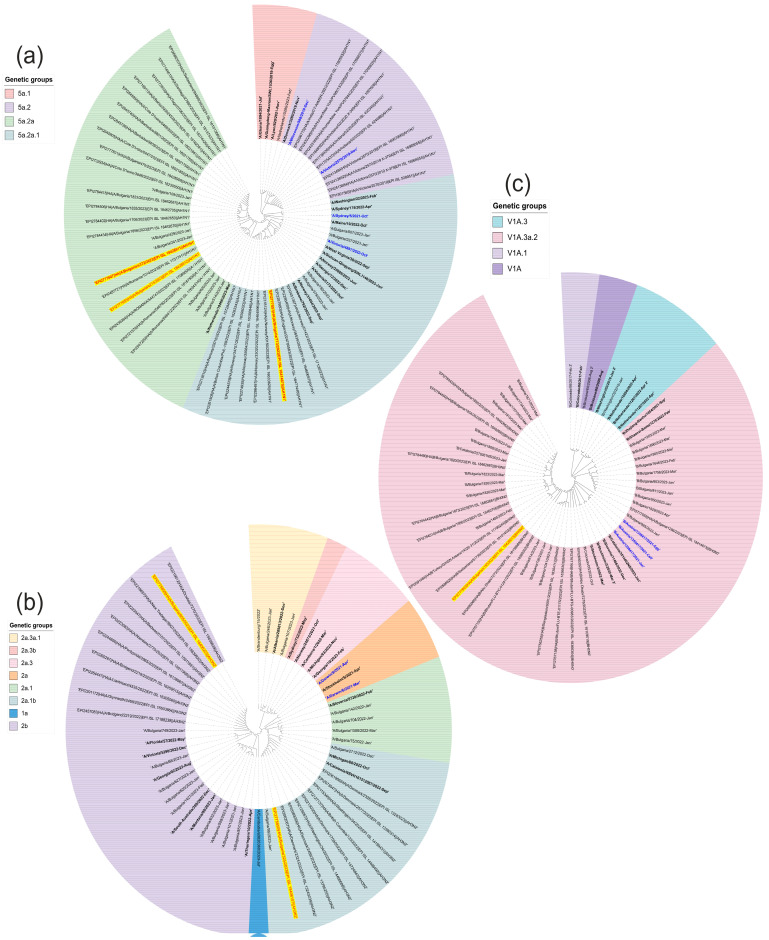
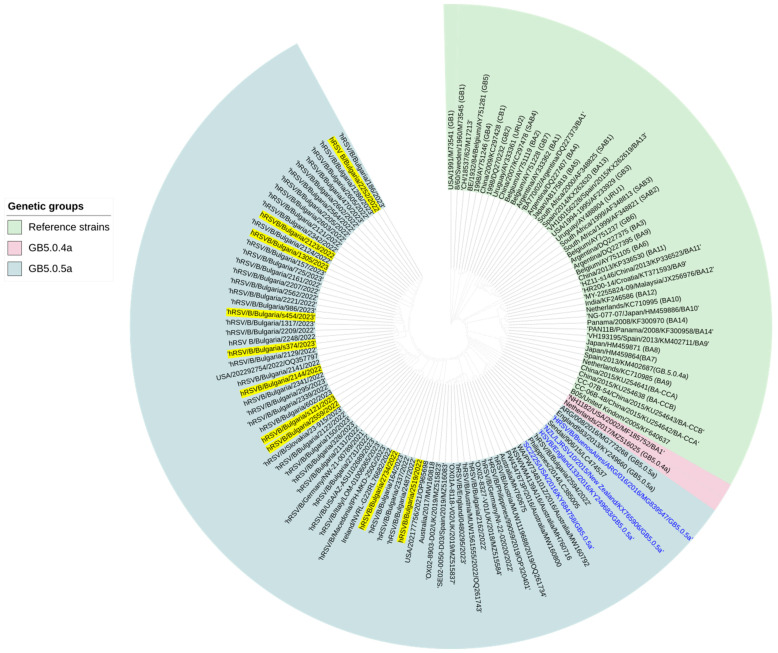
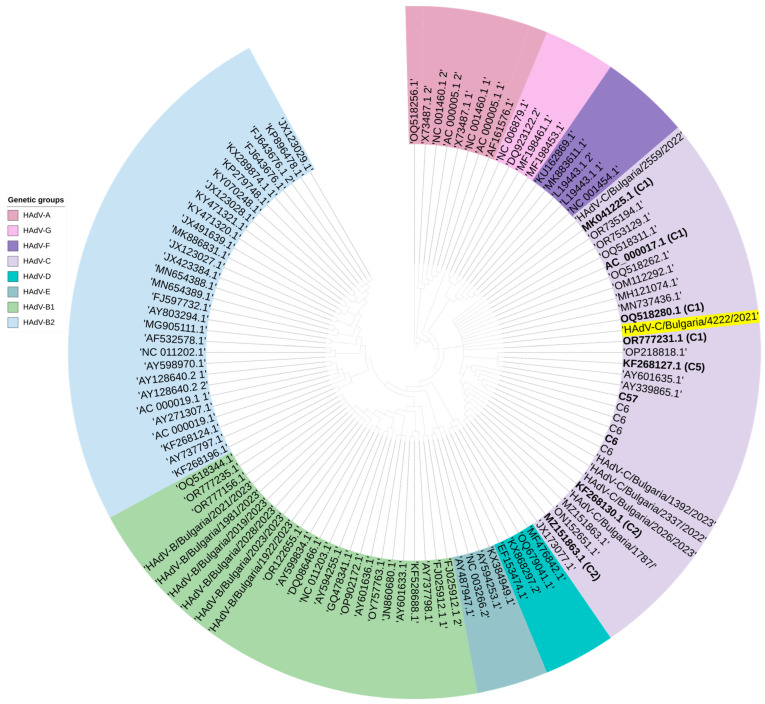
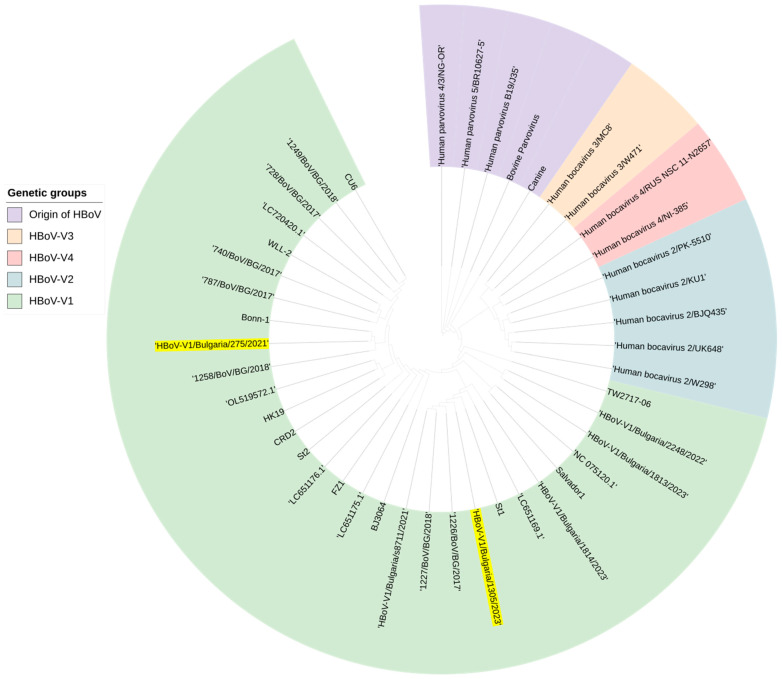
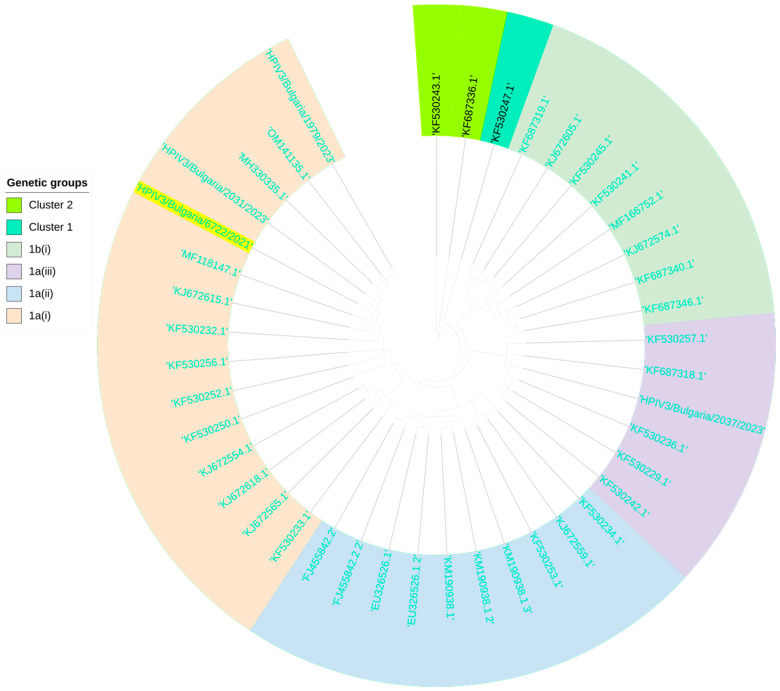
This study aimed to determine the incidence and etiological, seasonal, and genetic characteristics of respiratory viral coinfections involving severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Between October 2020 and January 2024, nasopharyngeal samples were collected from 2277 SARS-CoV-2-positive patients. Two multiplex approaches were used to detect and sequence SARS-CoV-2, influenza A/B viruses, and other seasonal respiratory viruses: multiplex real-time polymerase chain reaction (PCR) and multiplex next-generation sequencing. Coinfections of SARS-CoV-2 with other respiratory viruses were detected in 164 (7.2%) patients. The most common co-infecting virus was respiratory syncytial virus (RSV) (38 cases, 1.7%), followed by bocavirus (BoV) (1.2%) and rhinovirus (RV) (1.1%). Patients ≤ 16 years of age had the highest rate (15%) of mixed infections. Whole-genome sequencing produced 19 complete genomes of seasonal respiratory viral co-pathogens, which were subjected to phylogenetic and amino acid analyses. The detected influenza viruses were classified into the genetic groups 6B.1A.5a.2a and 6B.1A.5a.2a.1 for A(H1N1)pdm09, 3C.2a1b.2a.2a.1 and 3C.2a.2b for A(H3N2), and V1A.3a.2 for the B/Victoria lineage. The RSV-B sequences belonged to the genetic group GB5.0.5a, with HAdV-C belonging to type 1, BoV to genotype VP1, and PIV3 to lineage 1a(i). Multiple amino acid substitutions were identified, including at the antibody-binding sites. This study provides insights into respiratory viral coinfections involving SARS-CoV-2 and reinforces the importance of genetic characterization of co-pathogens in the development of therapeutic and preventive strategies.
Keywords: AdV-C; BoV; PIV3; RSV-B; SARS-CoV-2; coinfections; influenza viruses; multiplex NGS; respiratory viruses.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



Similar articles
-
Resurgence of influenza with increased genetic diversity of circulating viruses during the 2022-2023 season.J Med Microbiol. 2024 Jul;73(7). doi: 10.1099/jmm.0.001864. J Med Microbiol. 2024. PMID: 39073070
-
[Simultaneous detection of respiratory viruses and influenza A virus subtypes using multiplex PCR].Mikrobiyol Bul. 2014 Oct;48(4):652-60. doi: 10.5578/mb.8221. Mikrobiyol Bul. 2014. PMID: 25492660 Turkish.
-
Low circulation of Influenza A and coinfection with SARS-CoV-2 among other respiratory viruses during the COVID-19 pandemic in a region of southern Brazil.J Med Virol. 2021 Jul;93(7):4392-4398. doi: 10.1002/jmv.26975. Epub 2021 Apr 8. J Med Virol. 2021. PMID: 33829531 Free PMC article.
-
Assessing respiratory viral exclusion and affinity interactions through co-infection incidence in a pediatric population during the 2022 resurgence of influenza and RSV.Front Cell Infect Microbiol. 2023 Jun 14;13:1208235. doi: 10.3389/fcimb.2023.1208235. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37389220 Free PMC article. Review.
-
Altered epidemiological patterns of Respiratory Syncytial Virus and influenza detections in a tropical Australian setting 2020 to 2023.Aust N Z J Public Health. 2024 Aug;48(4):100172. doi: 10.1016/j.anzjph.2024.100172. Epub 2024 Jul 25. Aust N Z J Public Health. 2024. PMID: 39059095 Review.
Cited by
-
Bacterial and Viral Co-Infections in COVID-19 Patients: Etiology and Clinical Impact.Biomedicines. 2024 Sep 27;12(10):2210. doi: 10.3390/biomedicines12102210. Biomedicines. 2024. PMID: 39457522 Free PMC article.
References
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
