

DNA methylation variations underlie lettuce domestication and divergence
- PMID: 38886807
- PMCID: PMC11184767
- DOI: 10.1186/s13059-024-03310-x
DNA methylation variations underlie lettuce domestication and divergence
Abstract
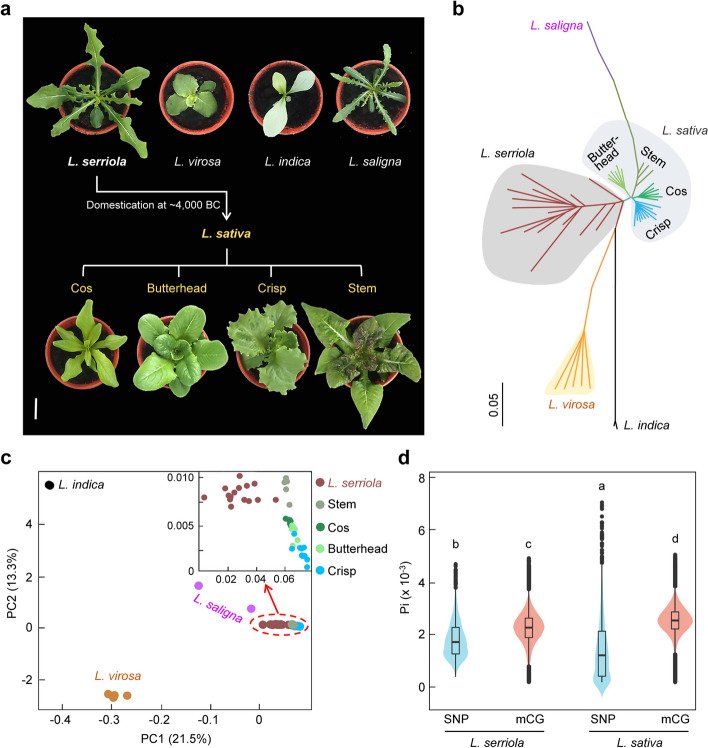
Background: Lettuce (Lactuca sativa L.) is an economically important vegetable crop worldwide. Lettuce is believed to be domesticated from a single wild ancestor Lactuca serriola and subsequently diverged into two major morphologically distinct vegetable types: leafy lettuce and stem lettuce. However, the role of epigenetic variation in lettuce domestication and divergence remains largely unknown.
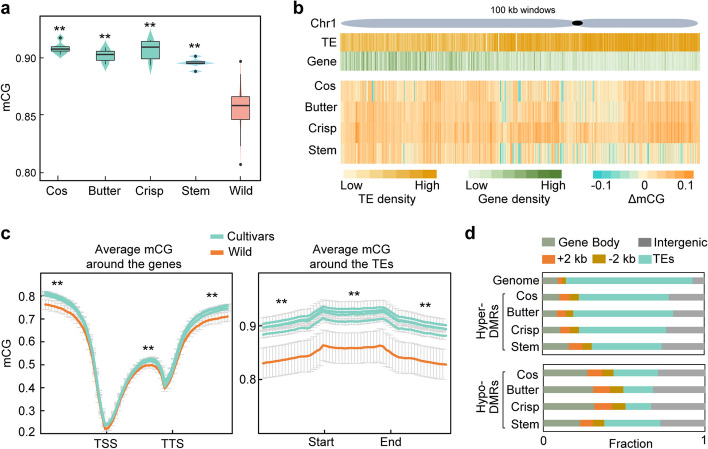
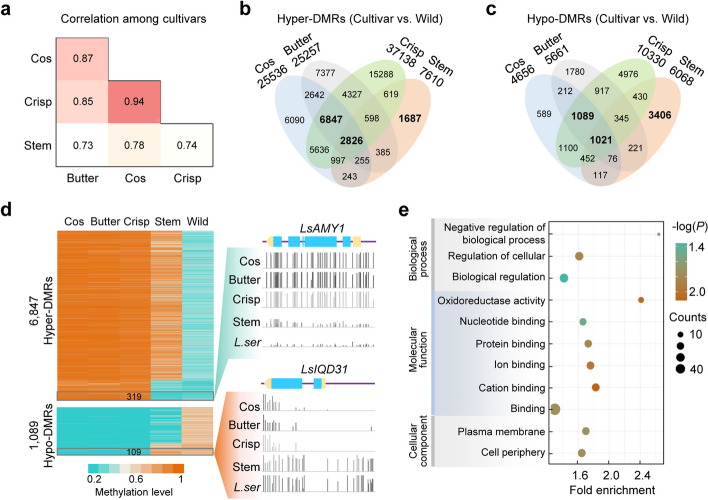
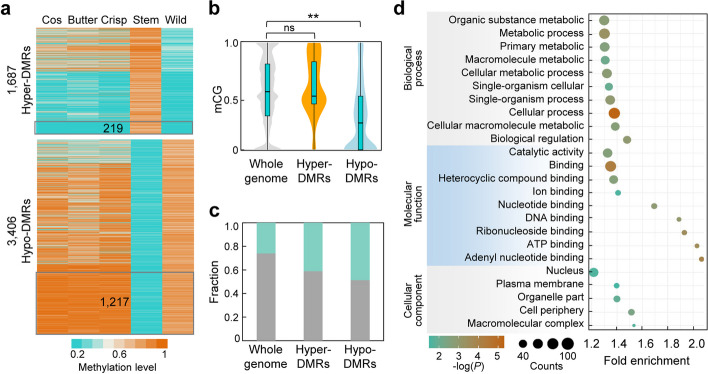
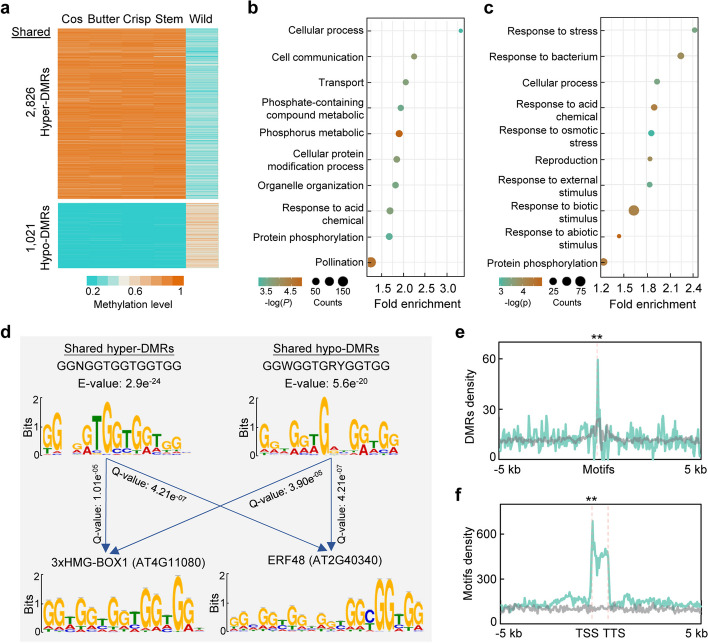
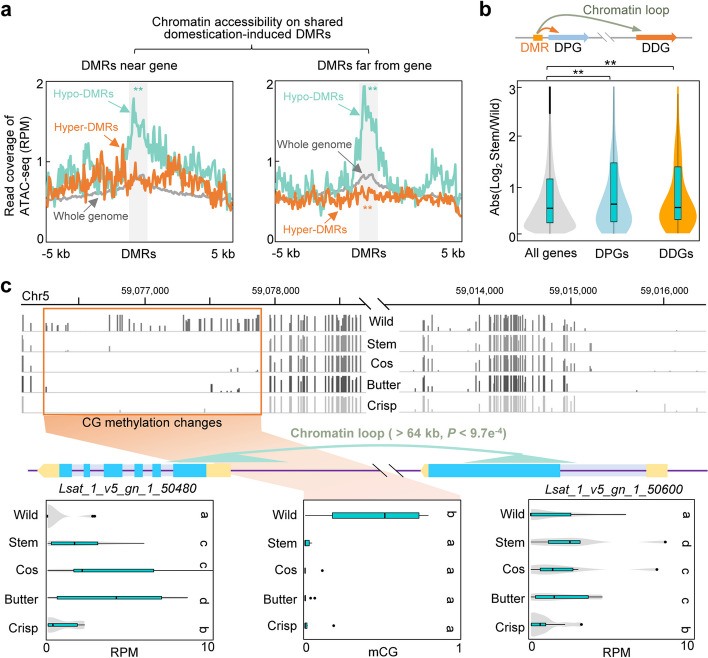
Results: To understand the genetic and epigenetic basis underlying lettuce domestication and divergence, we generate single-base resolution DNA methylomes from 52 Lactuca accessions, including major lettuce cultivars and wild relatives. We find a significant increase of DNA methylation during lettuce domestication and uncover abundant epigenetic variations associated with lettuce domestication and divergence. Interestingly, DNA methylation variations specifically associated with leafy and stem lettuce are related to regulation and metabolic processes, respectively, while those associated with both types are enriched in stress responses. Moreover, we reveal that domestication-induced DNA methylation changes could influence expression levels of nearby and distal genes possibly through affecting chromatin accessibility and chromatin loop.
Conclusion: Our study provides population epigenomic insights into crop domestication and divergence and valuable resources for further domestication for diversity and epigenetic breeding to boost crop improvement.
Keywords: DNA methylation; Divergence; Domestication; Epigenetic variation; Lettuce.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Whole-genome resequencing of 445 Lactuca accessions reveals the domestication history of cultivated lettuce.Nat Genet. 2021 May;53(5):752-760. doi: 10.1038/s41588-021-00831-0. Epub 2021 Apr 12. Nat Genet. 2021. PMID: 33846635
-
Dissection of the domestication-shaped genetic architecture of lettuce primary metabolism.Plant J. 2020 Nov;104(3):613-630. doi: 10.1111/tpj.14950. Epub 2020 Sep 14. Plant J. 2020. PMID: 32772408
-
Gapless genome assembly and epigenetic profiles reveal gene regulation of whole-genome triplication in lettuce.Gigascience. 2024 Jan 2;13:giae043. doi: 10.1093/gigascience/giae043. Gigascience. 2024. PMID: 38991853 Free PMC article.
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
Transgenerational epigenetic inheritance during plant evolution and breeding.Trends Plant Sci. 2024 Nov;29(11):1203-1223. doi: 10.1016/j.tplants.2024.04.007. Epub 2024 May 28. Trends Plant Sci. 2024. PMID: 38806375 Review.
References
-
- Shatilov MV, Razin AF, Ivanova MI. Analysis of the world lettuce market. IOP Conference Series. 2019;395:012053.
-
- Lebeda A, Ryder EJ, Grube R, Dolezˇalova´ I, Krˇ´ıstkova E. Lettuce (Asteraceae; Lactuca spp.). Genetic Resources, Chromosome Engineering, and Crop Improvement Vol 3 (ed Singh, R J). 2006:377-472.
-
- de Vries IM. Origin and domestication of Lactuca sativa L. Genet Resour Crop Ev. 1997;44:165–174. doi: 10.1023/A:1008611200727. - DOI
-
- Lindqvist K. On the origin of cultivated lettuce. Hereditas. 1960;46:319–350. doi: 10.1111/j.1601-5223.1960.tb03091.x. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
