

Diagnosis and management of non-CAH 46,XX disorders/differences in sex development
- PMID: 38812815
- PMCID: PMC11134272
- DOI: 10.3389/fendo.2024.1354759
Diagnosis and management of non-CAH 46,XX disorders/differences in sex development
Abstract
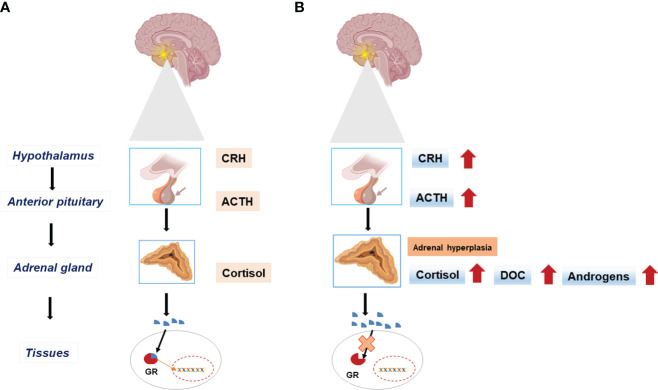
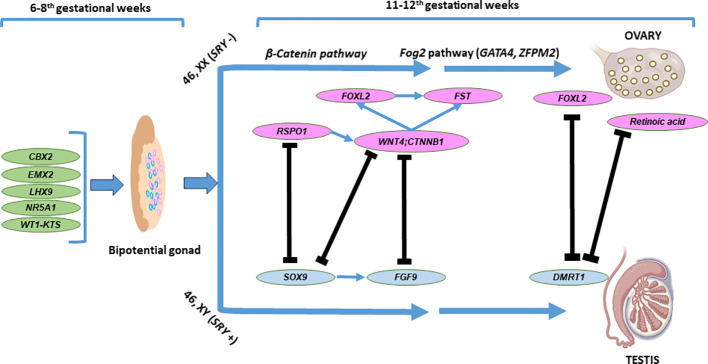
Prenatal-onset androgen excess leads to abnormal sexual development in 46,XX individuals. This androgen excess can be caused endogenously by the adrenals or gonads or by exposure to exogenous androgens. The most common cause of 46,XX disorders/differences in sex development (DSD) is congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, comprising >90% of 46,XX DSD cases. Deficiencies of 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase, and P450-oxidoreductase (POR) are rare types of CAH, resulting in 46,XX DSD. In all CAH forms, patients have normal ovarian development. The molecular genetic causes of 46,XX DSD, besides CAH, are uncommon. These etiologies include primary glucocorticoid resistance (PGCR) and aromatase deficiency with normal ovarian development. Additionally, 46,XX gonads can differentiate into testes, causing 46,XX testicular (T) DSD or a coexistence of ovarian and testicular tissue, defined as 46,XX ovotesticular (OT)-DSD. PGCR is caused by inactivating variants in NR3C1, resulting in glucocorticoid insensitivity and the signs of mineralocorticoid and androgen excess. Pathogenic variants in the CYP19A1 gene lead to aromatase deficiency, causing androgen excess. Many genes are involved in the mechanisms of gonadal development, and genes associated with 46,XX T/OT-DSD include translocations of the SRY; copy number variants in NR2F2, NR0B1, SOX3, SOX9, SOX10, and FGF9, and sequence variants in NR5A1, NR2F2, RSPO1, SOX9, WNT2B, WNT4, and WT1. Progress in cytogenetic and molecular genetic techniques has significantly improved our understanding of the etiology of non-CAH 46,XX DSD. Nonetheless, uncertainties about gonadal function and gender outcomes may make the management of these conditions challenging. This review explores the intricate landscape of diagnosing and managing these conditions, shedding light on the unique aspects that distinguish them from other types of DSD.
Keywords: DSD; aromatase deficiency; disorders/differences in sex development; gonadal dysgenesis; non-CAH 46, XX DSD; primary glucocorticoid resistance; testicular/ovotesticular disorders/differences in sex development.
Copyright © 2024 Yavas Abalı and Guran.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Molecular diagnostics of disorders of sexual development: an Indian survey and systems biology perspective.Syst Biol Reprod Med. 2019 Apr;65(2):105-120. doi: 10.1080/19396368.2018.1549619. Epub 2018 Dec 14. Syst Biol Reprod Med. 2019. PMID: 30550360 Review.
-
Nonsyndromic 46,XX Testicular Disorders/Differences of Sex Development.2003 Oct 30 [updated 2022 May 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. 2003 Oct 30 [updated 2022 May 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 20301589 Free Books & Documents. Review.
-
46,XX DSD due to Androgen Excess in Monogenic Disorders of Steroidogenesis: Genetic, Biochemical, and Clinical Features.Int J Mol Sci. 2019 Sep 17;20(18):4605. doi: 10.3390/ijms20184605. Int J Mol Sci. 2019. PMID: 31533357 Free PMC article. Review.
-
Clinical review of 95 patients with 46,XX disorders of sex development based on the new Chicago classification.J Pediatr Adolesc Gynecol. 2015 Feb;28(1):6-11. doi: 10.1016/j.jpag.2014.01.106. Epub 2014 Nov 12. J Pediatr Adolesc Gynecol. 2015. PMID: 25444050
-
Are NR5A1 Variations a Frequent Cause of 46,XX Ovotesticular Disorders of Sex Development? Analysis from a Single Center and Systematic Review.Sex Dev. 2022;16(4):242-251. doi: 10.1159/000526036. Epub 2023 Jan 19. Sex Dev. 2022. PMID: 36657429
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials