

The gluconeogenesis enzyme PCK2 has a non-enzymatic role in proteostasis in endothelial cells
- PMID: 38783087
- PMCID: PMC11116505
- DOI: 10.1038/s42003-024-06186-6
The gluconeogenesis enzyme PCK2 has a non-enzymatic role in proteostasis in endothelial cells
Abstract
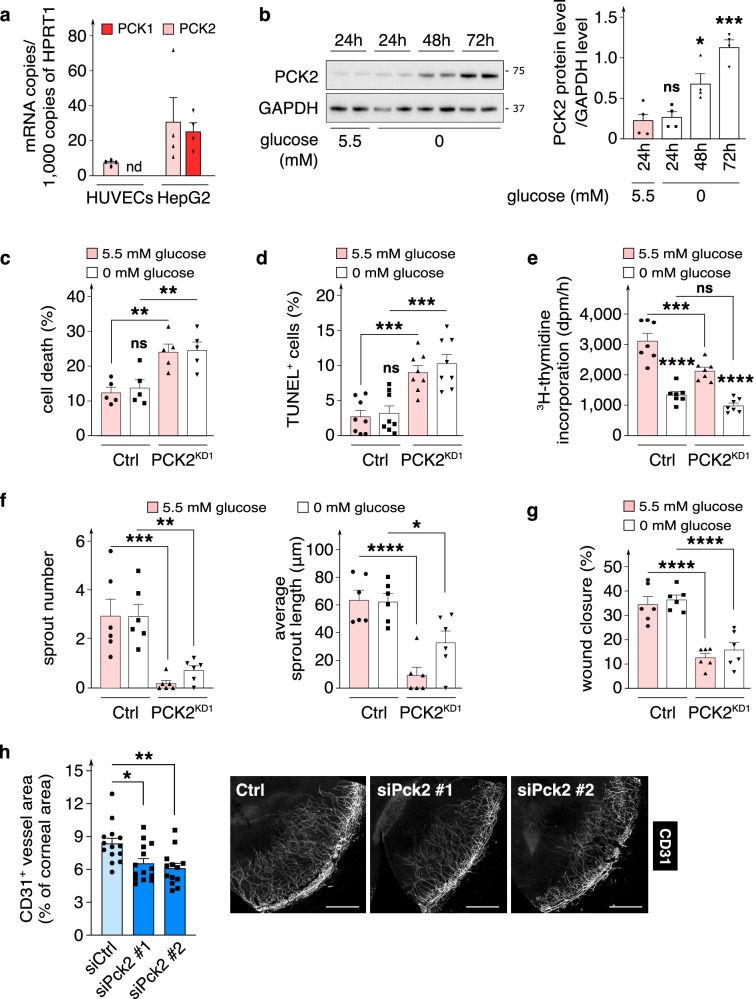
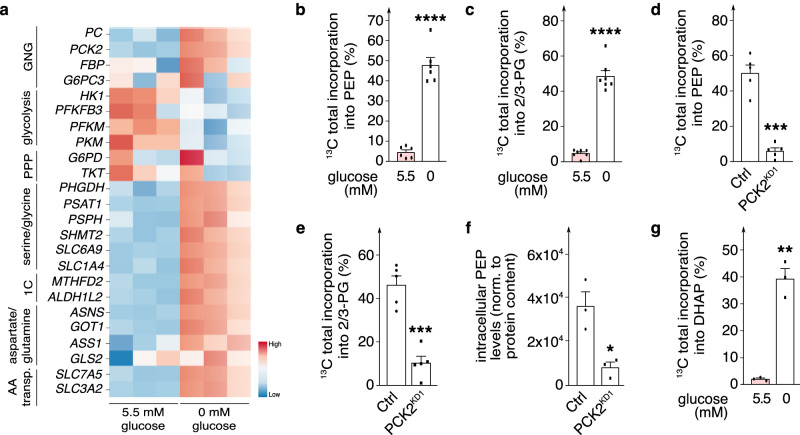
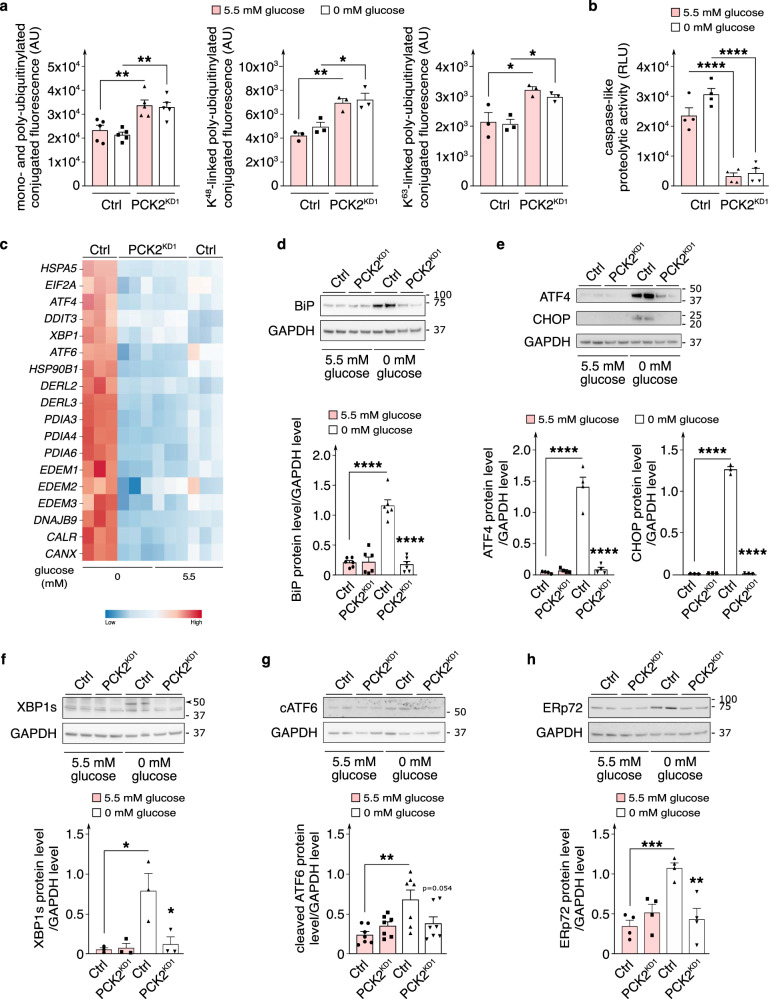
Endothelial cells (ECs) are highly glycolytic, but whether they generate glycolytic intermediates via gluconeogenesis (GNG) in glucose-deprived conditions remains unknown. Here, we report that glucose-deprived ECs upregulate the GNG enzyme PCK2 and rely on a PCK2-dependent truncated GNG, whereby lactate and glutamine are used for the synthesis of lower glycolytic intermediates that enter the serine and glycerophospholipid biosynthesis pathways, which can play key roles in redox homeostasis and phospholipid synthesis, respectively. Unexpectedly, however, even in normal glucose conditions, and independent of its enzymatic activity, PCK2 silencing perturbs proteostasis, beyond its traditional GNG role. Indeed, PCK2-silenced ECs have an impaired unfolded protein response, leading to accumulation of misfolded proteins, which due to defective proteasomes and impaired autophagy, results in the accumulation of protein aggregates in lysosomes and EC demise. Ultimately, loss of PCK2 in ECs impaired vessel sprouting. This study identifies a role for PCK2 in proteostasis beyond GNG.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability.J Biol Chem. 2014 Aug 8;289(32):22090-102. doi: 10.1074/jbc.M114.566927. Epub 2014 Jun 27. J Biol Chem. 2014. PMID: 24973213 Free PMC article.
-
Gluconeogenesis in cancer cells - Repurposing of a starvation-induced metabolic pathway?Biochim Biophys Acta Rev Cancer. 2019 Aug;1872(1):24-36. doi: 10.1016/j.bbcan.2019.05.006. Epub 2019 May 30. Biochim Biophys Acta Rev Cancer. 2019. PMID: 31152822 Free PMC article. Review.
-
PCK2 opposes mitochondrial respiration and maintains the redox balance in starved lung cancer cells.Free Radic Biol Med. 2021 Nov 20;176:34-45. doi: 10.1016/j.freeradbiomed.2021.09.007. Epub 2021 Sep 11. Free Radic Biol Med. 2021. PMID: 34520823
-
Biallelic pathogenic variants in the mitochondrial form of phosphoenolpyruvate carboxykinase cause peripheral neuropathy.HGG Adv. 2023 Jan 21;4(2):100182. doi: 10.1016/j.xhgg.2023.100182. eCollection 2023 Apr 13. HGG Adv. 2023. PMID: 36845668 Free PMC article.
-
Intricate Regulation of Phosphoenolpyruvate Carboxykinase (PEPCK) Isoforms in Normal Physiology and Disease.Curr Mol Med. 2019;19(4):247-272. doi: 10.2174/1566524019666190404155801. Curr Mol Med. 2019. PMID: 30947672 Review.
References
-
- Kalucka, J. et al. Quiescent endothelial cells upregulate fatty acid beta-oxidation for vasculoprotection via redox homeostasis. CellMetab. 28, 881–894 (2018). - PubMed
-
- Vandekeere, S. et al. Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 28, 573–587 (2018). - PubMed
MeSH terms
Substances
Grants and funding
- G.0B11.16N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- PhD Fellowship/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- Postdoctoral fellowship/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- Postdoctoral fellowship/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
LinkOut - more resources
Full Text Sources
Research Materials
