

Therapeutic developments for neurodegenerative GM1 gangliosidosis
- PMID: 38737101
- PMCID: PMC11082364
- DOI: 10.3389/fnins.2024.1392683
Therapeutic developments for neurodegenerative GM1 gangliosidosis
Abstract
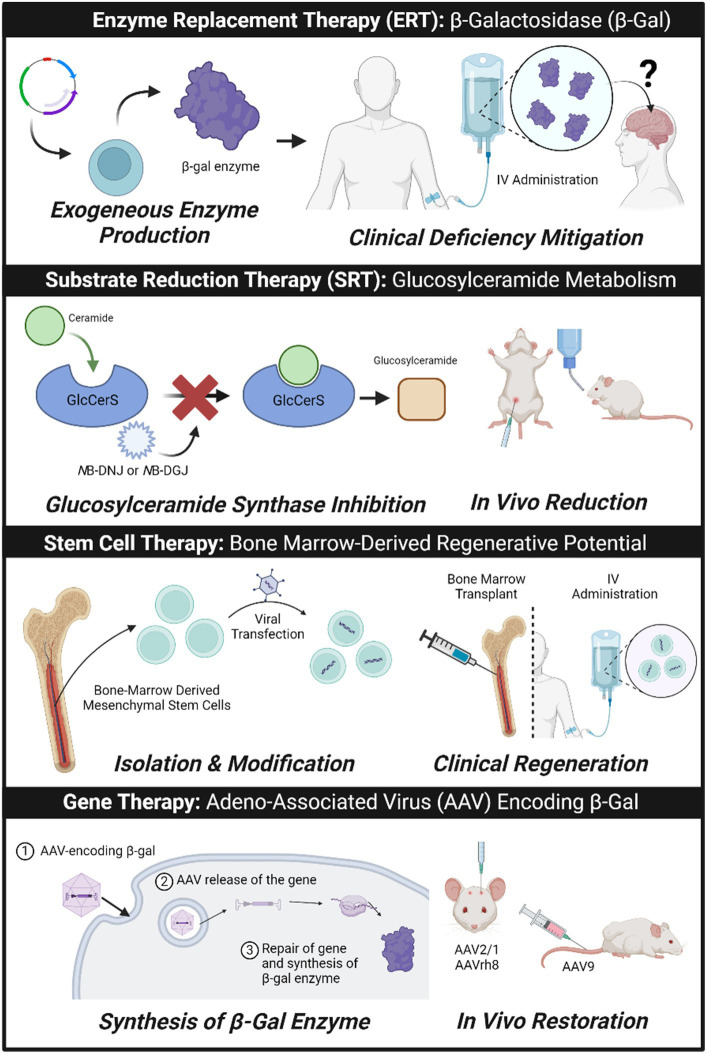
GM1 gangliosidosis (GM1) is a rare but fatal neurodegenerative disease caused by dysfunction or lack of production of lysosomal enzyme, β-galactosidase, leading to accumulation of substrates. The most promising treatments for GM1, include enzyme replacement therapy (ERT), substrate reduction therapy (SRT), stem cell therapy and gene editing. However, effectiveness is limited for neuropathic GM1 due to the restrictive nature of the blood-brain barrier (BBB). ERT and SRT alleviate substrate accumulation through exogenous supplementation over the patient's lifetime, while gene editing could be curative, fixing the causative gene, GLB1, to enable endogenous enzyme activity. Stem cell therapy can be a combination of both, with ex vivo gene editing of cells to cause the production of enzymes. These approaches require special considerations for brain delivery, which has led to novel formulations. A few therapeutic interventions have progressed to early-phase clinical trials, presenting a bright outlook for improved clinical management for GM1.
Keywords: GM1 gangliosidosis; clinical trials; enzyme replacement therapy; gene therapy; lysosomal storage disease; neurodegeneration; substrate reduction therapy.
Copyright © 2024 Foster, Williams, Arnold and Larsen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Intracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice.J Biol Chem. 2020 Sep 25;295(39):13532-13555. doi: 10.1074/jbc.RA119.009811. Epub 2019 Sep 3. J Biol Chem. 2020. PMID: 31481471 Free PMC article.
-
Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis.Cells. 2022 Aug 19;11(16):2579. doi: 10.3390/cells11162579. Cells. 2022. PMID: 36010656 Free PMC article.
-
GM1 Gangliosidosis: Mechanisms and Management.Appl Clin Genet. 2021 Apr 9;14:209-233. doi: 10.2147/TACG.S206076. eCollection 2021. Appl Clin Genet. 2021. PMID: 33859490 Free PMC article. Review.
-
Enzyme replacement for GM1-gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase:RTB lectin fusion.Mol Genet Metab. 2016 Feb;117(2):199-209. doi: 10.1016/j.ymgme.2015.12.002. Epub 2015 Dec 8. Mol Genet Metab. 2016. PMID: 26766614 Free PMC article.
-
Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease.Brain Dev. 2013 Jun;35(6):515-23. doi: 10.1016/j.braindev.2012.12.002. Epub 2013 Jan 3. Brain Dev. 2013. PMID: 23290321 Review.
Cited by
-
Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model.Int J Mol Sci. 2024 Sep 8;25(17):9712. doi: 10.3390/ijms25179712. Int J Mol Sci. 2024. PMID: 39273659 Free PMC article.
-
Differential Tractography: A Biomarker for Neuronal Function in Neurodegenerative Disease.medRxiv [Preprint]. 2024 Aug 26:2024.08.25.24312255. doi: 10.1101/2024.08.25.24312255. medRxiv. 2024. PMID: 39371116 Free PMC article. Preprint.
References
-
- Andersson U., Smith D., Jeyakumar M., Butters T. D., Borja M. C., Dwek R. A., et al. . (2004). Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol. Dis. 16, 506–515. doi: 10.1016/j.nbd.2004.04.012, PMID: - DOI - PubMed
-
- Broekman M. L. D., Baek R. C., Comer L. A., Fernandez J. L., Seyfried T. N., Sena-Esteves M. (2007). Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus-mediated gene delivery. Mol. Ther. 15, 30–37. doi: 10.1038/sj.mt.6300004, PMID: - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources