

m1A regulator-mediated methylation modification patterns correlated with autophagy to predict the prognosis of hepatocellular carcinoma
- PMID: 38649860
- PMCID: PMC11034060
- DOI: 10.1186/s12885-024-12235-4
m1A regulator-mediated methylation modification patterns correlated with autophagy to predict the prognosis of hepatocellular carcinoma
Abstract
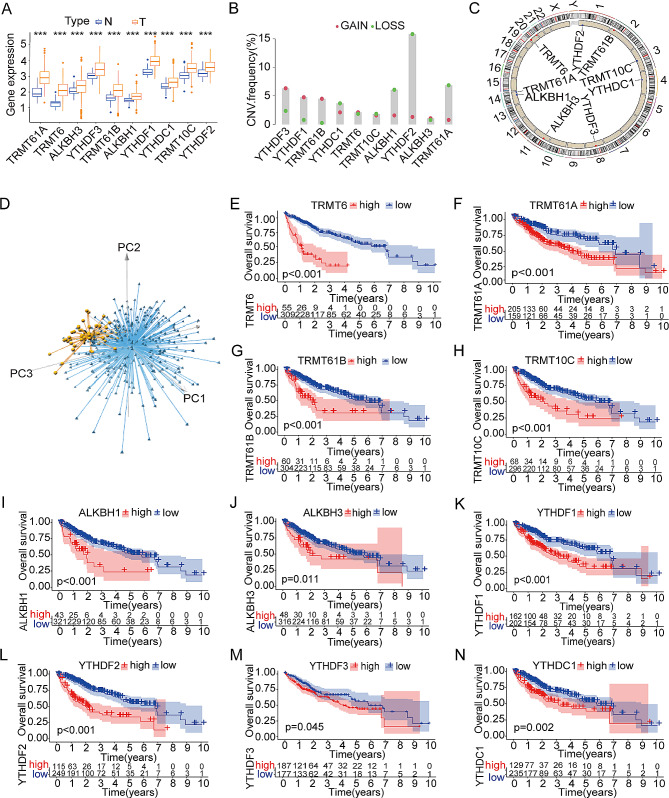
Background: N1-methyladenosine (m1A), among the most common internal modifications on RNAs, has a crucial role to play in cancer development. The purpose of this study were systematically investigate the modification characteristics of m1A in hepatocellular carcinoma (HCC) to unveil its potential as an anticancer target and to develop a model related to m1A modification characteristics with biological functions. This model could predict the prognosis for patients with HCC.
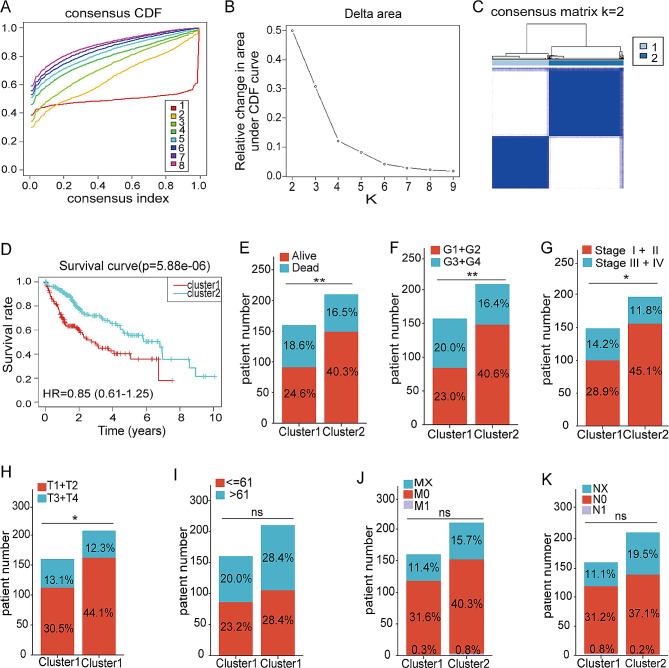
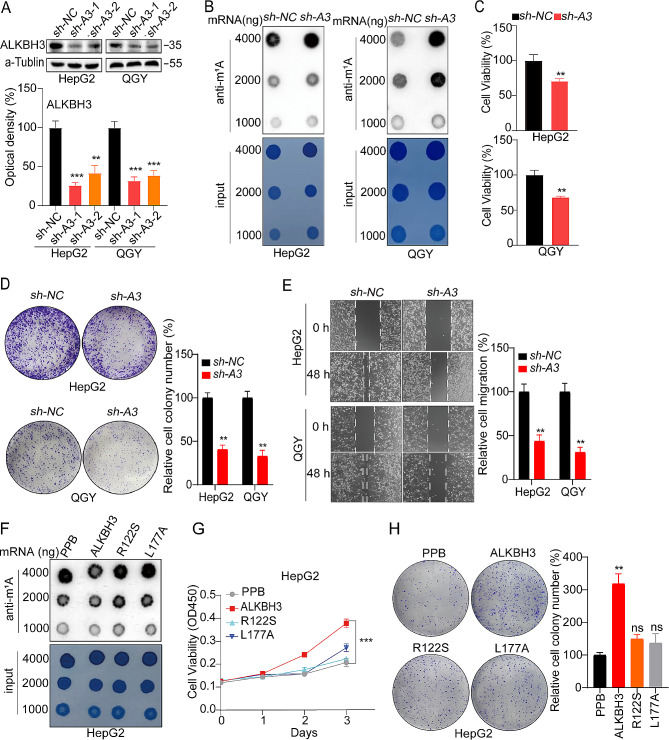
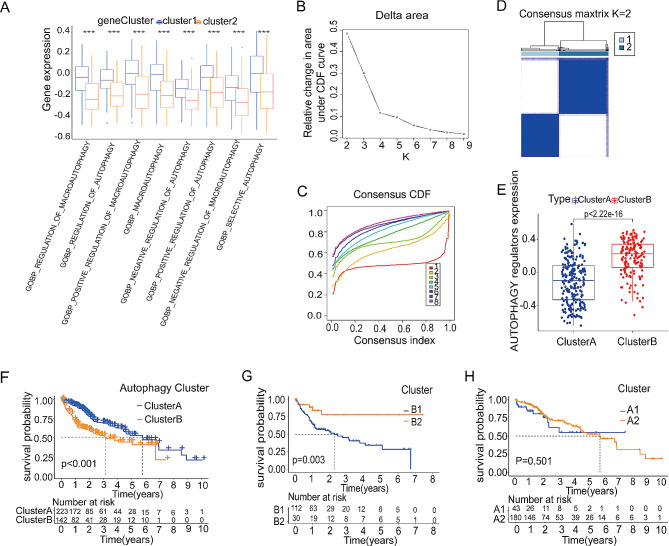
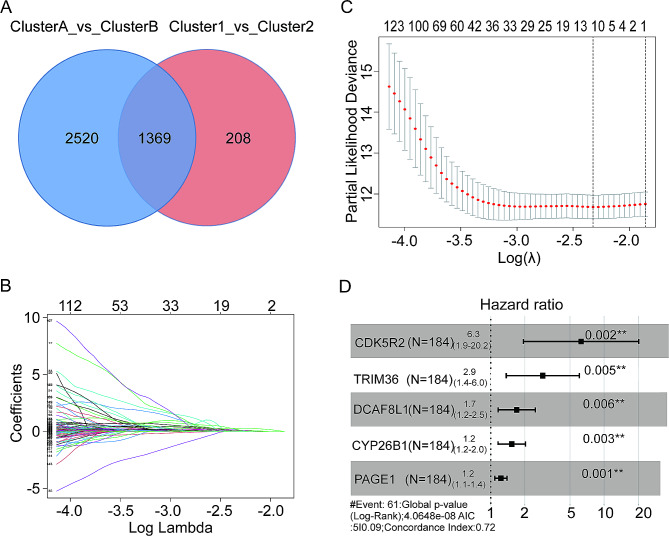
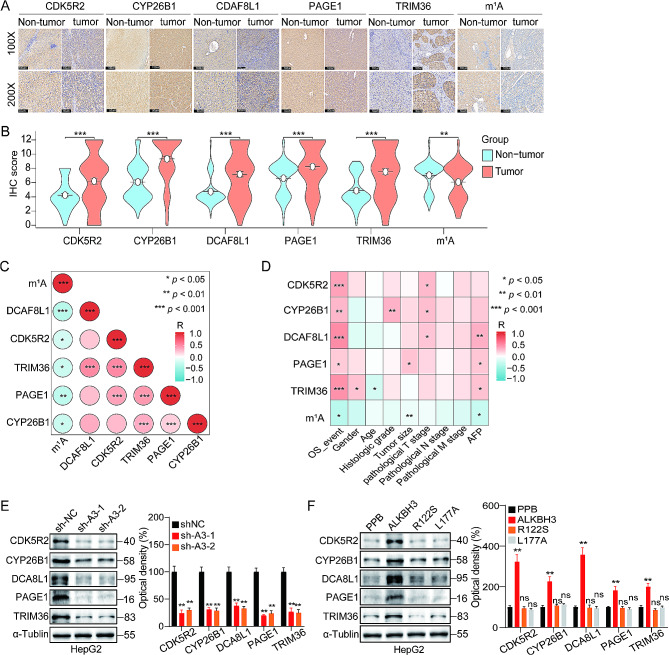
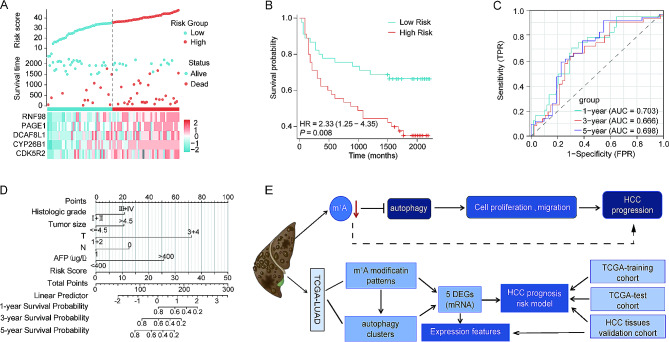
Methods: An integrated analysis of the TCGA-LIHC database was performed to explore the gene signatures and clinical relevance of 10 m1A regulators. Furthermore, the biological pathways regulated by m1A modification patterns were investigated. The risk model was established using the genes that showed differential expression (DEGs) between various m1A modification patterns and autophagy clusters. These in vitro experiments were subsequently designed to validate the role of m1A in HCC cell growth and autophagy. Immunohistochemistry was employed to assess m1A levels and the expression of DEGs from the risk model in HCC tissues and paracancer tissues using tissue microarray.
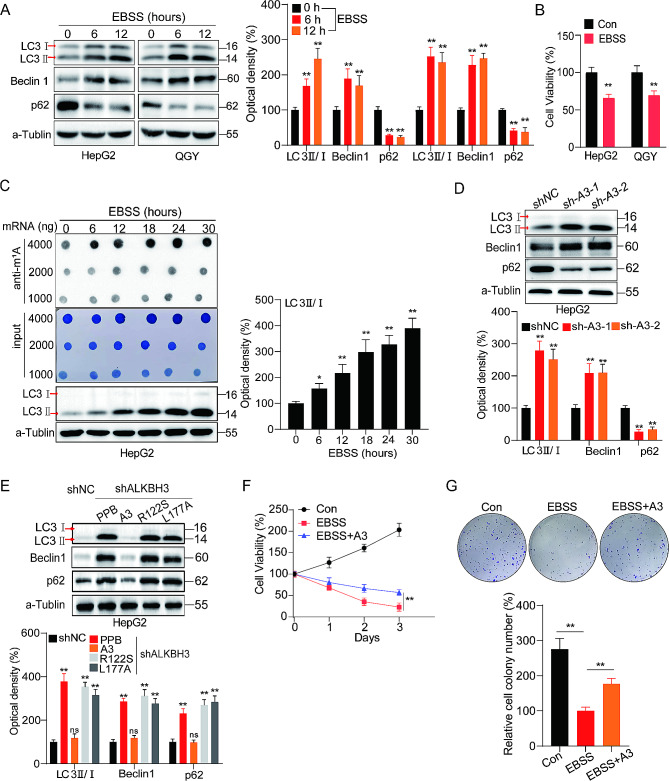
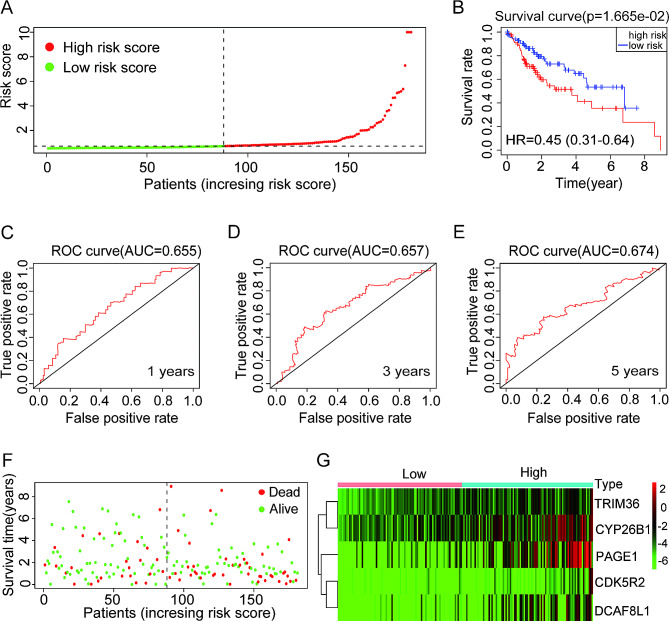
Results: The risk model, constructed from five DEGs (CDK5R2, TRIM36, DCAF8L, CYP26B, and PAGE1), exhibited significant prognostic value in predicting survival rates among individuals with HCC. Moreover, HCC tissues showed decreased levels of m1A compared to paracancer tissues. Furthermore, the low m1A level group indicated a poorer clinical outcome for patients with HCC. Additionally, m1A modification may positively influence autophagy regulation, thereby inhibiting HCC cells proliferation under nutrient deficiency conditions.
Conclusions: The risk model, comprising m1A regulators correlated with autophagy and constructed from five DEGs, could be instrumental in predicting HCC prognosis. The reduced level of m1A may represent a potential target for anti-HCC strategies.
Keywords: Autophagy; Hepatocellular carcinoma (HCC); N1-methyladenosine (m1A); Prognosis; m1A regulators.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no potential conflict of interest.
Figures

Similar articles
-
Expression pattern and prognostic value of N6-methyladenosine RNA methylation key regulators in hepatocellular carcinoma.Mutagenesis. 2021 Oct 6;36(5):369-379. doi: 10.1093/mutage/geab032. Mutagenesis. 2021. PMID: 34467992 Free PMC article.
-
m6A RNA Methylation Regulators Elicit Malignant Progression and Predict Clinical Outcome in Hepatocellular Carcinoma.Dis Markers. 2021 Jun 4;2021:8859590. doi: 10.1155/2021/8859590. eCollection 2021. Dis Markers. 2021. PMID: 34234878 Free PMC article.
-
A m6A regulators-related classifier for prognosis and tumor microenvironment characterization in hepatocellular carcinoma.Front Immunol. 2024 Jul 25;15:1374465. doi: 10.3389/fimmu.2024.1374465. eCollection 2024. Front Immunol. 2024. PMID: 39119345 Free PMC article.
-
The role of RNA modifications in hepatocellular carcinoma: functional mechanism and potential applications.Front Immunol. 2024 Aug 20;15:1439485. doi: 10.3389/fimmu.2024.1439485. eCollection 2024. Front Immunol. 2024. PMID: 39229278 Free PMC article. Review.
-
The functional roles, cross-talk and clinical implications of m6A modification and circRNA in hepatocellular carcinoma.Int J Biol Sci. 2021 Jul 22;17(12):3059-3079. doi: 10.7150/ijbs.62767. eCollection 2021. Int J Biol Sci. 2021. PMID: 34421350 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
- 82103296 and 82260606/The National Natural Science Foundation of China
- 2021M700971/The China Postdoctoral Science Foundation
- ZK[2022]390/The Guizhou Provincial Science and Technology Projects
- QJJ[2022]191/Guizhou Provincial Department of Education for Higher Education Scientific Research Foundation
- gzwkj2022-254/Guizhou Provincial Health Commission Science and Technology Foundation
LinkOut - more resources
Full Text Sources
