

Convergent epigenetic evolution drives relapse in acute myeloid leukemia
- PMID: 38647535
- PMCID: PMC11034943
- DOI: 10.7554/eLife.93019
Convergent epigenetic evolution drives relapse in acute myeloid leukemia
Abstract
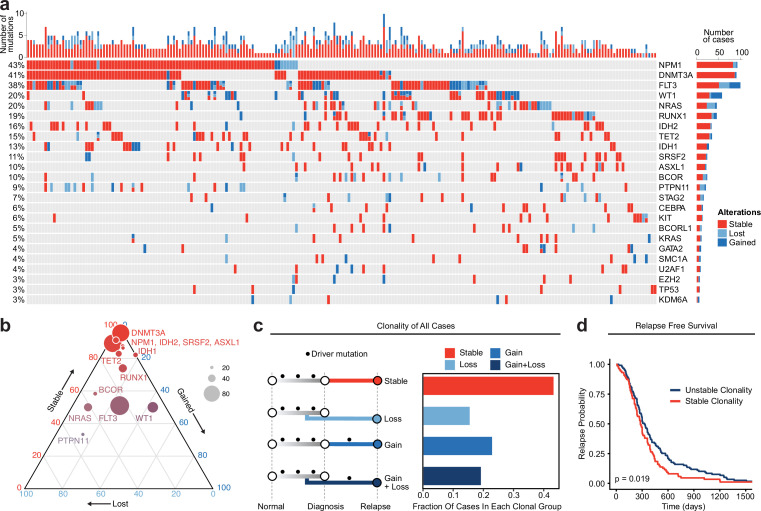
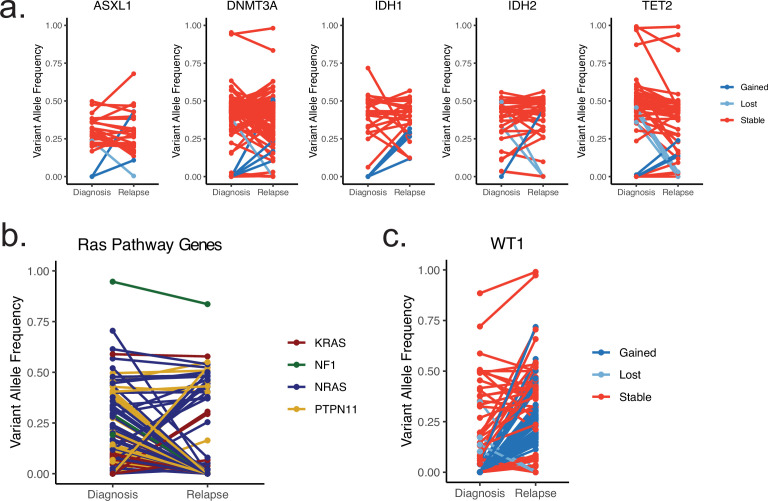
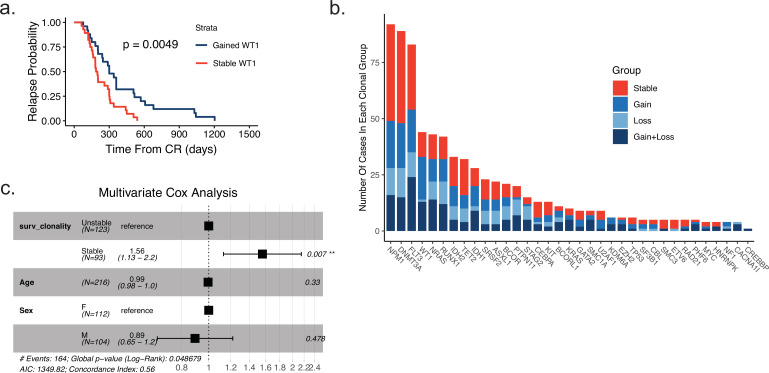
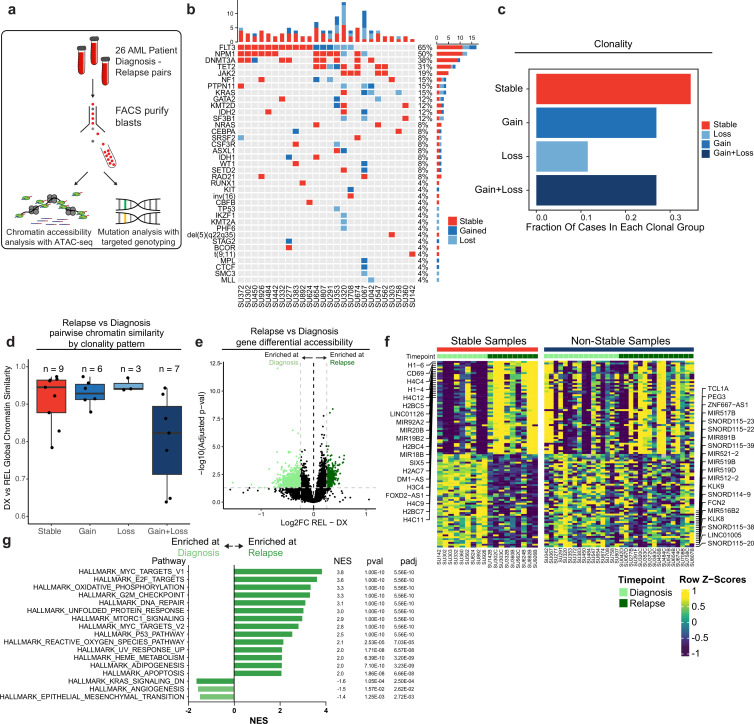
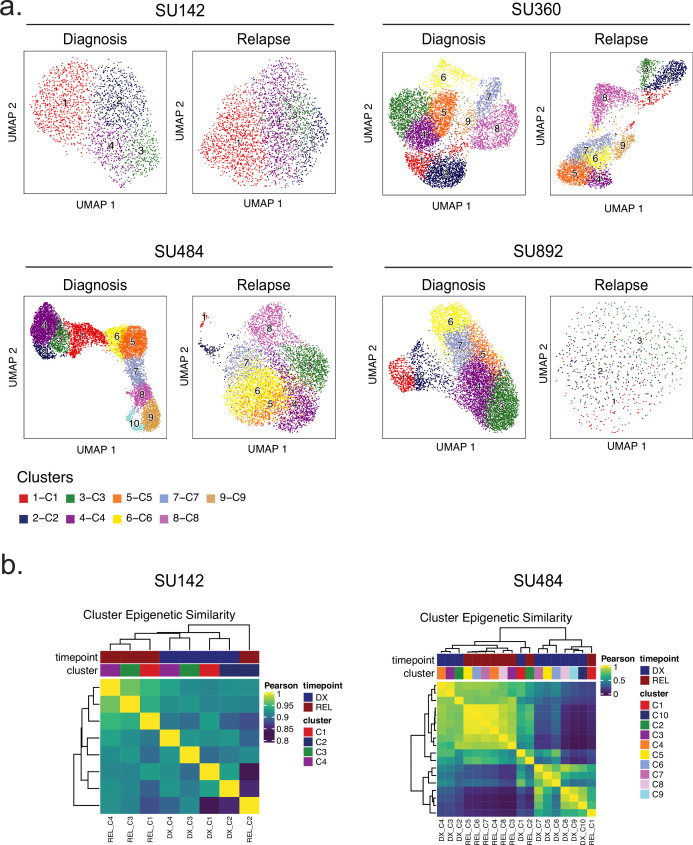
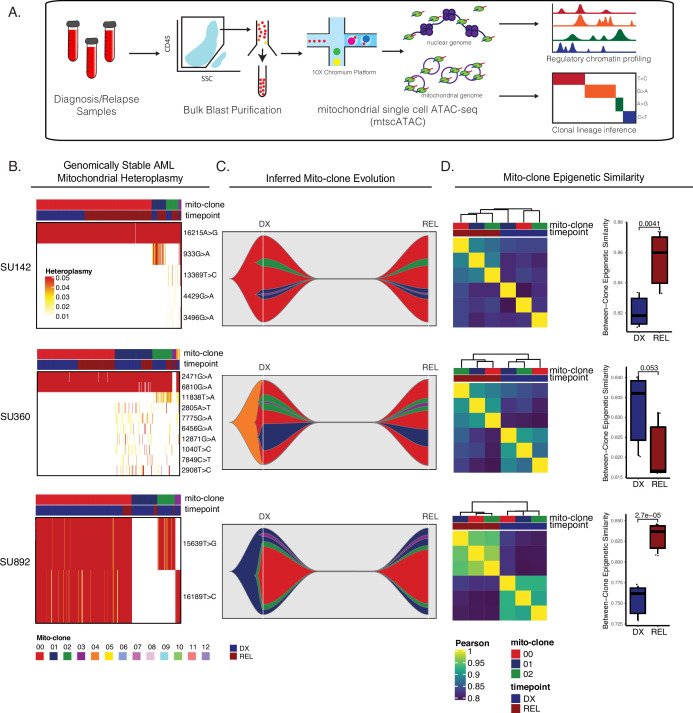
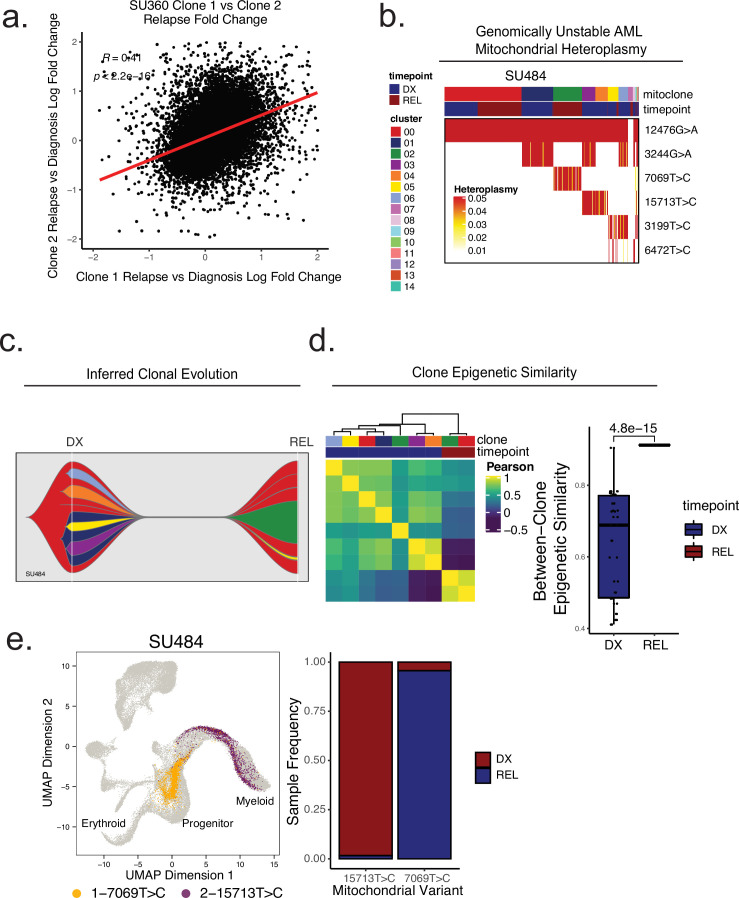
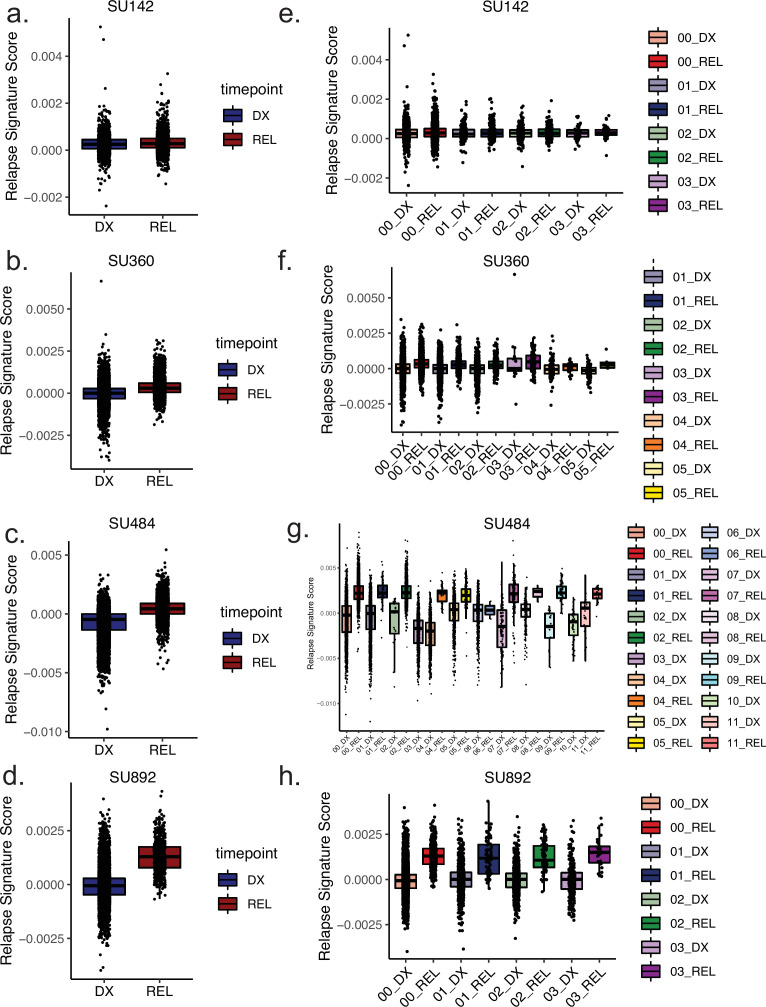
Relapse of acute myeloid leukemia (AML) is highly aggressive and often treatment refractory. We analyzed previously published AML relapse cohorts and found that 40% of relapses occur without changes in driver mutations, suggesting that non-genetic mechanisms drive relapse in a large proportion of cases. We therefore characterized epigenetic patterns of AML relapse using 26 matched diagnosis-relapse samples with ATAC-seq. This analysis identified a relapse-specific chromatin accessibility signature for mutationally stable AML, suggesting that AML undergoes epigenetic evolution at relapse independent of mutational changes. Analysis of leukemia stem cell (LSC) chromatin changes at relapse indicated that this leukemic compartment underwent significantly less epigenetic evolution than non-LSCs, while epigenetic changes in non-LSCs reflected overall evolution of the bulk leukemia. Finally, we used single-cell ATAC-seq paired with mitochondrial sequencing (mtscATAC) to map clones from diagnosis into relapse along with their epigenetic features. We found that distinct mitochondrially-defined clones exhibit more similar chromatin accessibility at relapse relative to diagnosis, demonstrating convergent epigenetic evolution in relapsed AML. These results demonstrate that epigenetic evolution is a feature of relapsed AML and that convergent epigenetic evolution can occur following treatment with induction chemotherapy.
Keywords: acute myeloid leukemia; cancer biology; epigenetics; genetics; genomics; human; relapse.
Plain language summary
Acute myeloid leukemia (or AML for short) is a type of blood cancer characterized by abnormally high production of immature white blood cells. Despite advances in AML treatment, many patients relapse after an initially successful first round of treatment. As a result, understanding the factors contributing to relapse is essential for developing effective treatments for the disease. Like most cancers, AML can evolve because of changes to the DNA sequence in cells that cause them to grow uncontrollably or resist treatment. Alongside these genetic mutations, AML cells also undergo ‘epigenetic’ changes, where regions of the DNA are modified and genes can be switched on or off without altering the DNA sequence. Previous research has demonstrated that epigenetic changes contribute to the development of AML, however, it was not clear if these changes could also make cells resistant to treatment without acquiring new DNA mutations. Nuno, Azizi et al. addressed this question by analyzing the epigenetic states of AML cells from 26 patients at the time of their diagnosis and after treatment when the disease had relapsed. Analysis revealed that almost half of the patients with AML experienced a relapse without acquiring new DNA mutations. Instead, these AML cells developed specific epigenetic changes that helped them to resist cancer treatment. Moreover, studying individual AML cells from different patients showed that the cells became more epigenetically similar at relapse, suggesting that they converge towards a more treatment-resistant disease. Future experiments will determine exactly how these epigenetic changes lead to treatment resistance. Currently, most of the drugs used to treat AML are either chemotherapies or ones that target specific DNA mutations. The findings of Nuno, Azizi et al. suggest that drugs targeting specific epigenetic changes may be more effective for some patients. Further studies will be needed to determine which patients may benefit and which epigenetic drugs could be useful.
© 2024, Nuno, Azizi et al.
Conflict of interest statement
KN, AA, TK, AE, MC No competing interests declared, CL consultant for Cartography Biosciences, AS founder of Immunai and Cartography Biosciences; receives research funding from Allogene Therapeutics and Merck Research Laboratories, RM on the Advisory Boards of Kodikaz Therapeutic Solutions, Orbital Therapeutics, and is an inventor on a number of patents related to CD47 cancer immunotherapy licensed to Gilead Sciences. Co-founder and equity holder of Pheast Therapeutics, MyeloGene, and Orbital Therapeutics
Figures


Update of
-
Convergent Epigenetic Evolution Drives Relapse in Acute Myeloid Leukemia.bioRxiv [Preprint]. 2023 Oct 10:2023.10.10.561642. doi: 10.1101/2023.10.10.561642. bioRxiv. 2023. Update in: Elife. 2024 Apr 22;13:e93019. doi: 10.7554/eLife.93019 PMID: 37873452 Free PMC article. Updated. Preprint.
Similar articles
-
Convergent Epigenetic Evolution Drives Relapse in Acute Myeloid Leukemia.bioRxiv [Preprint]. 2023 Oct 10:2023.10.10.561642. doi: 10.1101/2023.10.10.561642. bioRxiv. 2023. Update in: Elife. 2024 Apr 22;13:e93019. doi: 10.7554/eLife.93019 PMID: 37873452 Free PMC article. Updated. Preprint.
-
Blast cells surviving acute myeloid leukemia induction therapy are in cycle with a signature of FOXM1 activity.BMC Cancer. 2021 Oct 28;21(1):1153. doi: 10.1186/s12885-021-08839-9. BMC Cancer. 2021. PMID: 34711181 Free PMC article.
-
Loss of Kat2a enhances transcriptional noise and depletes acute myeloid leukemia stem-like cells.Elife. 2020 Jan 27;9:e51754. doi: 10.7554/eLife.51754. Elife. 2020. PMID: 31985402 Free PMC article.
-
Exploiting epigenetically mediated changes: Acute myeloid leukemia, leukemia stem cells and the bone marrow microenvironment.Adv Cancer Res. 2019;141:213-253. doi: 10.1016/bs.acr.2018.12.005. Epub 2019 Jan 21. Adv Cancer Res. 2019. PMID: 30691684 Review.
-
The contributing factors of resistance or sensitivity to epigenetic drugs in the treatment of AML.Clin Transl Oncol. 2022 Jul;24(7):1250-1261. doi: 10.1007/s12094-022-02776-0. Epub 2022 Jan 25. Clin Transl Oncol. 2022. PMID: 35076883 Review.
Cited by
-
Traditional Chinese medicine for acute myelocytic leukemia therapy: exploiting epigenetic targets.Front Pharmacol. 2024 Jun 4;15:1388903. doi: 10.3389/fphar.2024.1388903. eCollection 2024. Front Pharmacol. 2024. PMID: 38895633 Free PMC article. Review.
-
Divergent Processing of Cell Stress Signals as the Basis of Cancer Progression: Licensing NFκB on Chromatin.Int J Mol Sci. 2024 Aug 7;25(16):8621. doi: 10.3390/ijms25168621. Int J Mol Sci. 2024. PMID: 39201306 Free PMC article. Review.
-
Current insights and future directions of LncRNA Morrbid in disease pathogenesis.Heliyon. 2024 Aug 22;10(17):e36681. doi: 10.1016/j.heliyon.2024.e36681. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39263145 Free PMC article. Review.
-
MOCHA's advanced statistical modeling of scATAC-seq data enables functional genomic inference in large human cohorts.Nat Commun. 2024 Aug 9;15(1):6828. doi: 10.1038/s41467-024-50612-6. Nat Commun. 2024. PMID: 39122670 Free PMC article.
-
Multi-omic analysis of longitudinal acute myeloid leukemia patient samples reveals potential prognostic markers linked to disease progression.Front Genet. 2024 Sep 27;15:1442539. doi: 10.3389/fgene.2024.1442539. eCollection 2024. Front Genet. 2024. PMID: 39399221 Free PMC article.
References
-
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, Perna F, Zhao X, Taylor JE, Park CY, Carroll M, Melnick A, Nimer SD, Jaffe JD, Aifantis I, Bernstein BE, Levine RL. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–193. doi: 10.1016/j.ccr.2012.06.032. - DOI - PMC - PubMed
-
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, Baboud J, Balasundaram M, Balu S, Barnabas N, Bartlett J, Bartlett P, Bastian BC, Baylin SB, Behera M, Belyaev D, Benz C, Bernard B, Beroukhim R, Bir N, Black AD, Bodenheimer T, Boice L, Boland GM, Bono R, Bootwalla MS, Bosenberg M, Bowen J, Bowlby R, Bristow CA, Brockway-Lunardi L, Brooks D, Brzezinski J, Bshara W, Buda E, Burns WR, Butterfield YSN, Button M, Calderone T, Cappellini GA, Carter C, Carter SL, Cherney L, Cherniack AD, Chevalier A, Chin L, Cho J, Cho RJ, Choi YL, Chu A, Chudamani S, Cibulskis K, Ciriello G, Clarke A, Coons S, Cope L, Crain D, Curley E, Danilova L, D’Atri S, Davidsen T, Davies MA, Delman KA, Demchok JA, Deng QA, Deribe YL, Dhalla N, Dhir R, DiCara D, Dinikin M, Dubina M, Ebrom JS, Egea S, Eley G, Engel J, Eschbacher JM, Fedosenko KV, Felau I, Fennell T, Ferguson ML, Fisher S, Flaherty KT, Frazer S, Frick J, Fulidou V, Gabriel SB, Gao J, Gardner J, Garraway LA, Gastier-Foster JM, Gaudioso C, Gehlenborg N, Genovese G, Gerken M, Gershenwald JE, Getz G, Gomez-Fernandez C, Gribbin T, Grimsby J, Gross B, Guin R, Gutschner T, Hadjipanayis A, Halaban R, Hanf B, Haussler D, Haydu LE, Hayes DN, Hayward NK, Heiman DI, Herbert L, Herman JG, Hersey P, Hoadley KA, Hodis E, Holt RA, Hoon DSB, Hoppough S, Hoyle AP, Huang FW, Huang M, Huang S, Hutter CM, Ibbs M, Iype L, Jacobsen A, Jakrot V, Janning A, Jeck WR, Jefferys SR, Jensen MA, Jones CD, Jones SJM, Ju Z, Kakavand H, Kang H, Kefford RF, Khuri FR, Kim J, Kirkwood JM, Klode J, Korkut A, Korski K, Krauthammer M, Kucherlapati R, Kwong LN, Kycler W, Ladanyi M, Lai PH, Laird PW, Lander E, Lawrence MS, Lazar AJ, Łaźniak R, Lee D, Lee JE, Lee J, Lee K, Lee S, Lee W, Leporowska E, Leraas KM, Li HI, Lichtenberg TM, Lichtenstein L, Lin P, Ling S, Liu J, Liu O, Liu W, Long GV, Lu Y, Ma S, Ma Y, Mackiewicz A, Mahadeshwar HS, Malke J, Mallery D, Manikhas GM, Mann GJ, Marra MA, Matejka B, Mayo M, Mehrabi S, Meng S, Meyerson M, Mieczkowski PA, Miller JP, Miller ML, Mills GB, Moiseenko F, Moore RA, Morris S, Morrison C, Morton D, Moschos S, Mose LE, Muller FL, Mungall AJ, Murawa D, Murawa P, Murray BA, Nezi L, Ng S, Nicholson D, Noble MS, Osunkoya A, Owonikoko TK, Ozenberger BA, Pagani E, Paklina OV, Pantazi A, Parfenov M, Parfitt J, Park PJ, Park WY, Parker JS, Passarelli F, Penny R, Perou CM, Pihl TD, Potapova O, Prieto VG, Protopopov A, Quinn MJ, Radenbaugh A, Rai K, Ramalingam SS, Raman AT, Ramirez NC, Ramirez R, Rao U, Rathmell WK, Ren X, Reynolds SM, Roach J, Robertson AG, Ross MI, Roszik J, Russo G, Saksena G, Saller C, Samuels Y, Sander C, Sander C, Sandusky G, Santoso N, Saul M, Saw RPM, Schadendorf D, Schein JE, Schultz N, Schumacher SE, Schwallier C, Scolyer RA, Seidman J, Sekhar PC, Sekhon HS, Senbabaoglu Y, Seth S, Shannon KF, Sharpe S, Sharpless NE, Shaw KRM, Shelton C, Shelton T, Shen R, Sheth M, Shi Y, Shiau CJ, Shmulevich I, Sica GL, Simons JV, Sinha R, Sipahimalani P, Sofia HJ, Soloway MG, Song X, Sougnez C, Spillane AJ, Spychała A, Stretch JR, Stuart J, Suchorska WM, Sucker A, Sumer SO, Sun Y, Synott M, Tabak B, Tabler TR, Tam A, Tan D, Tang J, Tarnuzzer R, Tarvin K, Tatka H, Taylor BS, Teresiak M, Thiessen N, Thompson JF, Thorne L, Thorsson V, Trent JM, Triche TJ, Tsai KY, Tsou P, Van Den Berg DJ, Van Allen EM, Veluvolu U, Verhaak RG, Voet D, Voronina O, Walter V, Walton JS, Wan Y, Wang Y, Wang Z, Waring S, Watson IR, Weinhold N, Weinstein JN, Weisenberger DJ, White P, Wilkerson MD, Wilmott JS, Wise L, Wiznerowicz M, Woodman SE, Wu CJ, Wu CC, Wu J, Wu Y, Xi R, Xu AW, Yang D, Yang L, Yang L, Zack TI, Zenklusen JC, Zhang H, Zhang J, Zhang W, Zhao X, Zhu J, Zhu K, Zimmer L, Zmuda E, Zou L. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. - DOI - PMC - PubMed
-
- Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, Chin PS, Blair H, Cauchy P, James SR, Zacarias-Cabeza J, Gilding LN, Beggs A, Clokie S, Loke JC, Jenkin P, Uddin A, Delwel R, Richards SJ, Raghavan M, Griffiths MJ, Heidenreich O, Cockerill PN, Bonifer C. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nature Genetics. 2019;51:151–162. doi: 10.1038/s41588-018-0270-1. - DOI - PMC - PubMed
-
- Azizi A. Azizi/Nuno eLife 2024. swh:1:rev:f82a1cb04d6292ecdcf35c9e9c6d692c0fab84b2Software Heritage. 2024 https://archive.softwareheritage.org/swh:1:dir:2cdb2d4fed638289596f0e0cc...
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
