

This is a preprint.
Harnessing molecular mechanism for precision medicine in dilated cardiomyopathy caused by a mutation in troponin T
- PMID: 38645235
- PMCID: PMC11030379
- DOI: 10.1101/2024.04.05.588306
Harnessing molecular mechanism for precision medicine in dilated cardiomyopathy caused by a mutation in troponin T
Abstract
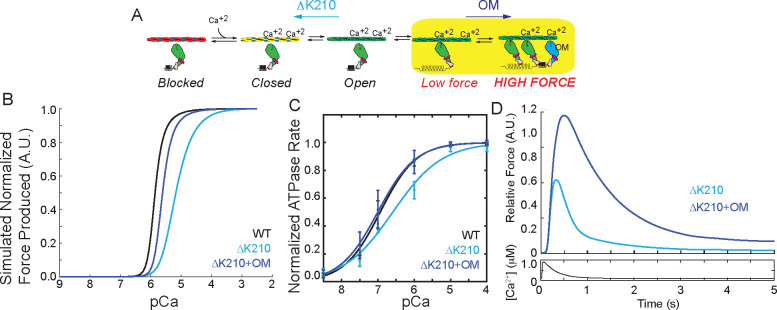
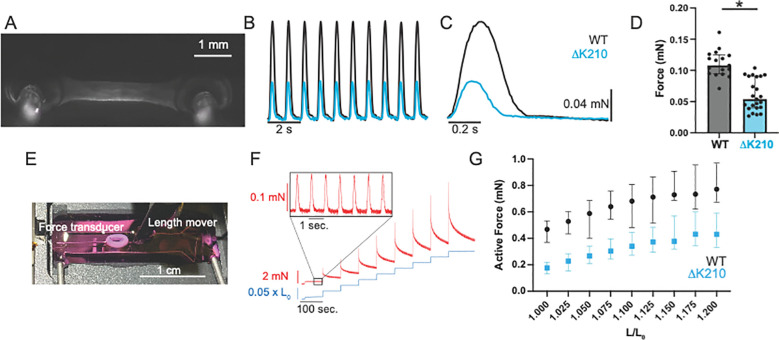
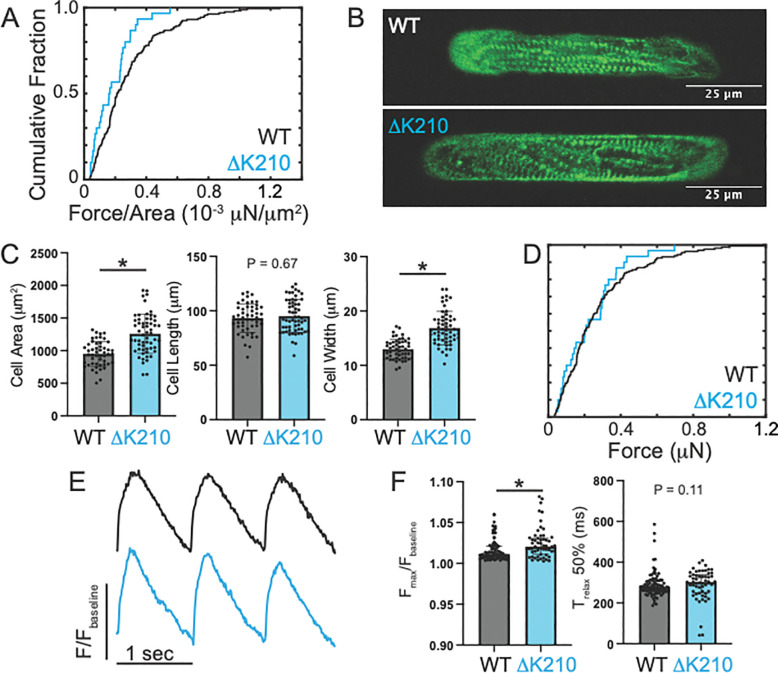
Familial dilated cardiomyopathy (DCM) is frequently caused by autosomal dominant point mutations in genes involved in diverse cellular processes, including sarcomeric contraction. While patient studies have defined the genetic landscape of DCM, genetics are not currently used in patient care, and patients receive similar treatments regardless of the underlying mutation. It has been suggested that a precision medicine approach based on the molecular mechanism of the underlying mutation could improve outcomes; however, realizing this approach has been challenging due to difficulties linking genotype and phenotype and then leveraging this information to identify therapeutic approaches. Here, we used multiscale experimental and computational approaches to test whether knowledge of molecular mechanism could be harnessed to connect genotype, phenotype, and drug response for a DCM mutation in troponin T, deletion of K210. Previously, we showed that at the molecular scale, the mutation reduces thin filament activation. Here, we used computational modeling of this molecular defect to predict that the mutant will reduce cellular and tissue contractility, and we validated this prediction in human cardiomyocytes and engineered heart tissues. We then used our knowledge of molecular mechanism to computationally model the effects of a small molecule that can activate the thin filament. We demonstrate experimentally that the modeling correctly predicts that the small molecule can partially rescue systolic dysfunction at the expense of diastolic function. Taken together, our results demonstrate how molecular mechanism can be harnessed to connect genotype and phenotype and inspire strategies to optimize mechanism-based therapeutics for DCM.
Keywords: Biological Sciences; Biophysics and Computational Biology; Troponin T; contractility; mechanobiology; myosin; stem cell derived cardiomyocytes.
Conflict of interest statement
Conflict of interest statement: All experiments were conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Figures



Similar articles
-
Dilated cardiomyopathy mutations in thin-filament regulatory proteins reduce contractility, suppress systolic Ca2+, and activate NFAT and Akt signaling.Am J Physiol Heart Circ Physiol. 2020 Aug 1;319(2):H306-H319. doi: 10.1152/ajpheart.00272.2020. Epub 2020 Jul 3. Am J Physiol Heart Circ Physiol. 2020. PMID: 32618513 Free PMC article.
-
Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca²⁺ sensitivity.Cardiovasc Res. 2013 Jul 1;99(1):65-73. doi: 10.1093/cvr/cvt071. Epub 2013 Mar 27. Cardiovasc Res. 2013. PMID: 23539503
-
Disrupted mechanobiology links the molecular and cellular phenotypes in familial dilated cardiomyopathy.Proc Natl Acad Sci U S A. 2019 Sep 3;116(36):17831-17840. doi: 10.1073/pnas.1910962116. Epub 2019 Aug 19. Proc Natl Acad Sci U S A. 2019. PMID: 31427533 Free PMC article.
-
Insights and Challenges of Multi-Scale Modeling of Sarcomere Mechanics in cTn and Tm DCM Mutants-Genotype to Cellular Phenotype.Front Physiol. 2017 Mar 14;8:151. doi: 10.3389/fphys.2017.00151. eCollection 2017. Front Physiol. 2017. PMID: 28352236 Free PMC article. Review.
-
[Genetics of dilated cardiomyopathy].Z Kardiol. 2001 Jul;90(7):461-9. doi: 10.1007/s003920170134. Z Kardiol. 2001. PMID: 11515275 Review. German.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources