

Impairment of the Glial Phagolysosomal System Drives Prion-Like Propagation in a Drosophila Model of Huntington's Disease
- PMID: 38589228
- PMCID: PMC11097281
- DOI: 10.1523/JNEUROSCI.1256-23.2024
Impairment of the Glial Phagolysosomal System Drives Prion-Like Propagation in a Drosophila Model of Huntington's Disease
Abstract
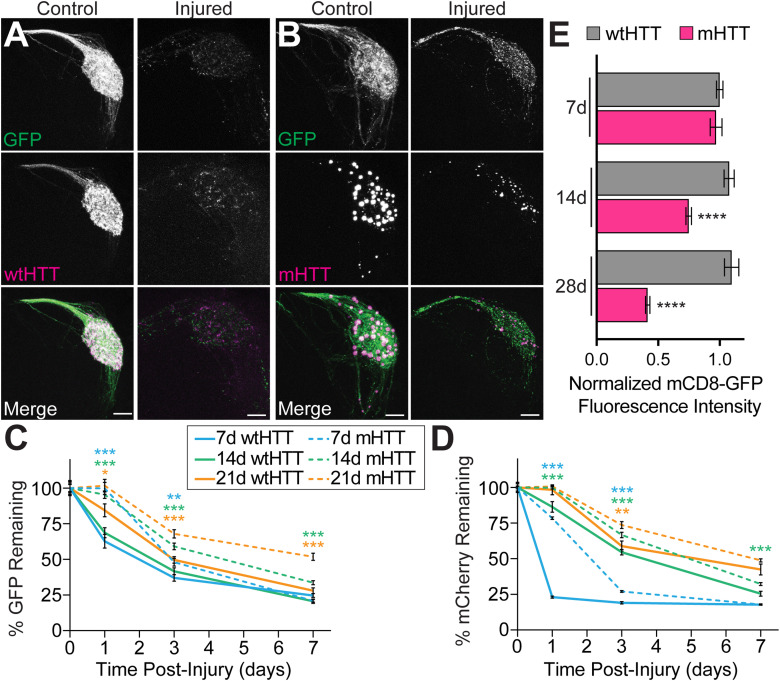
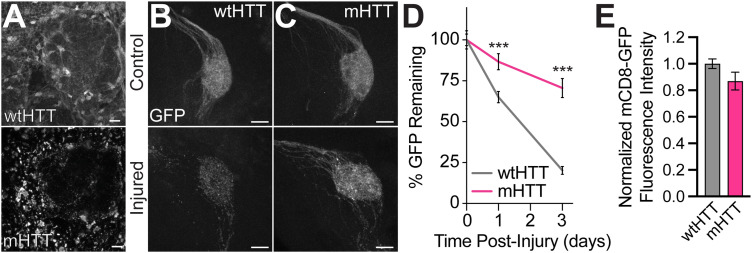
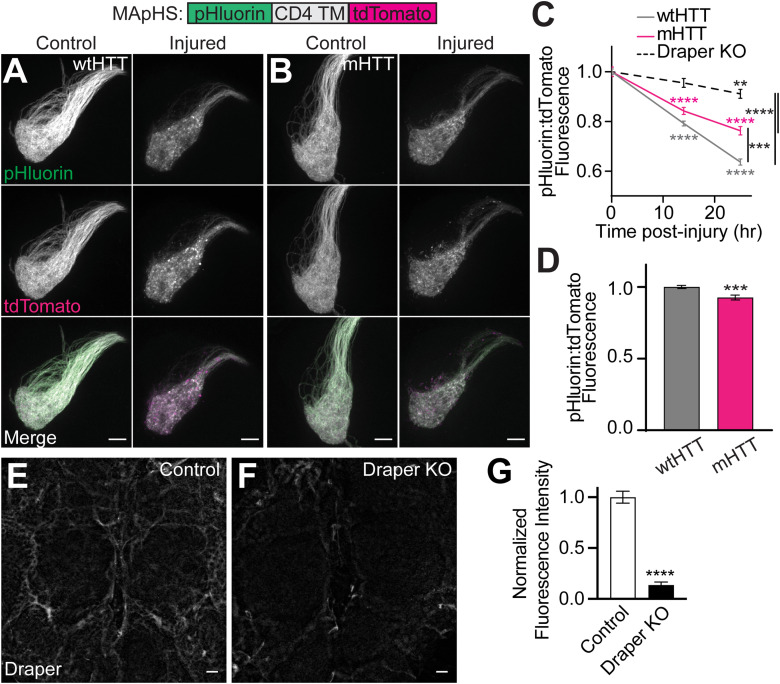
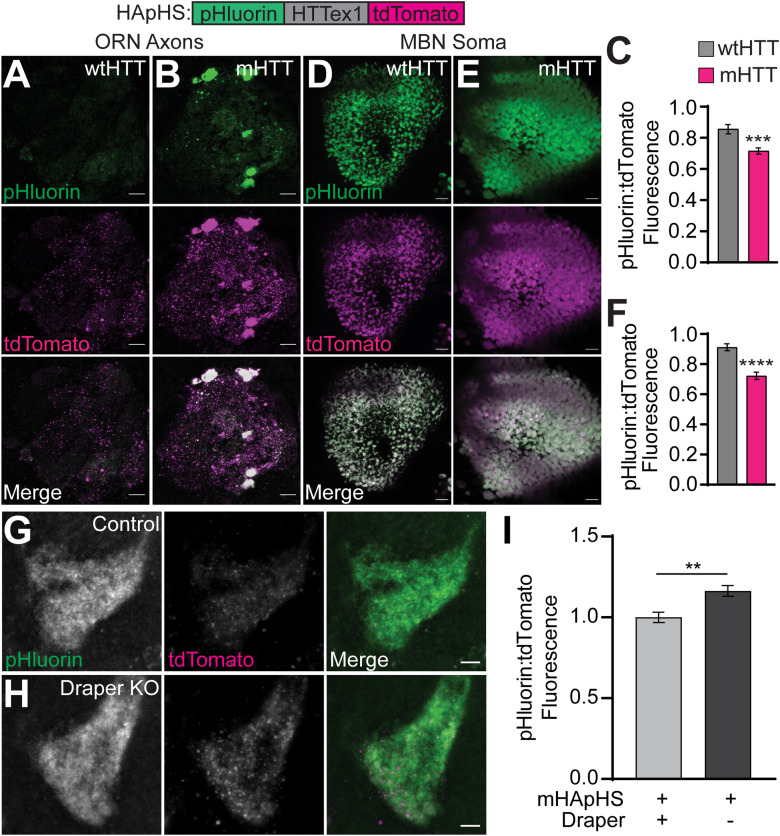
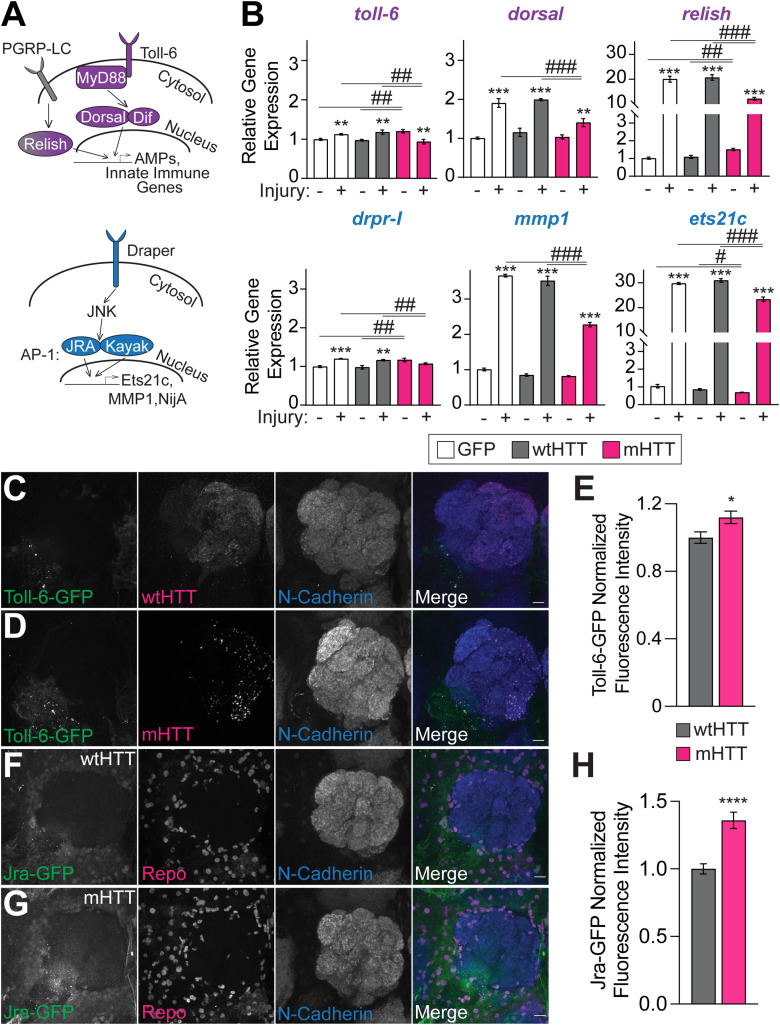
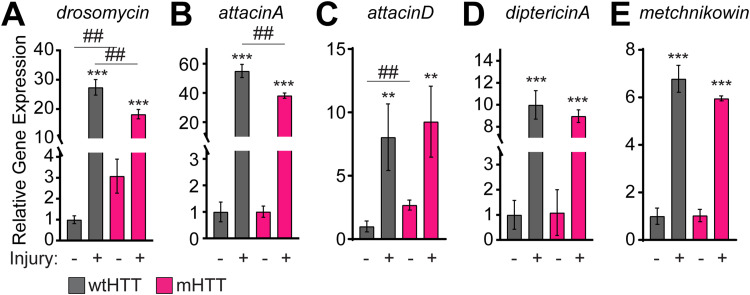
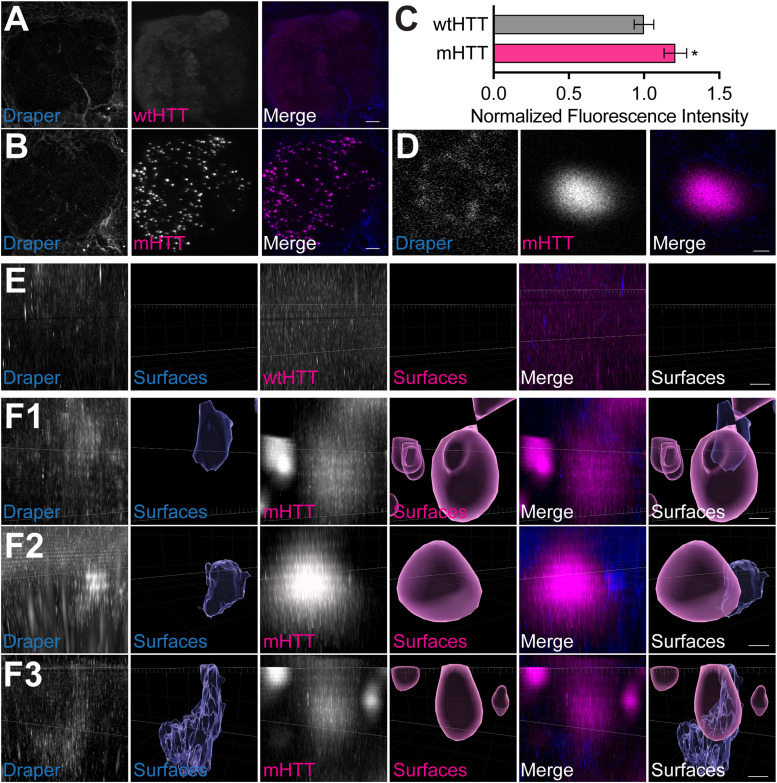
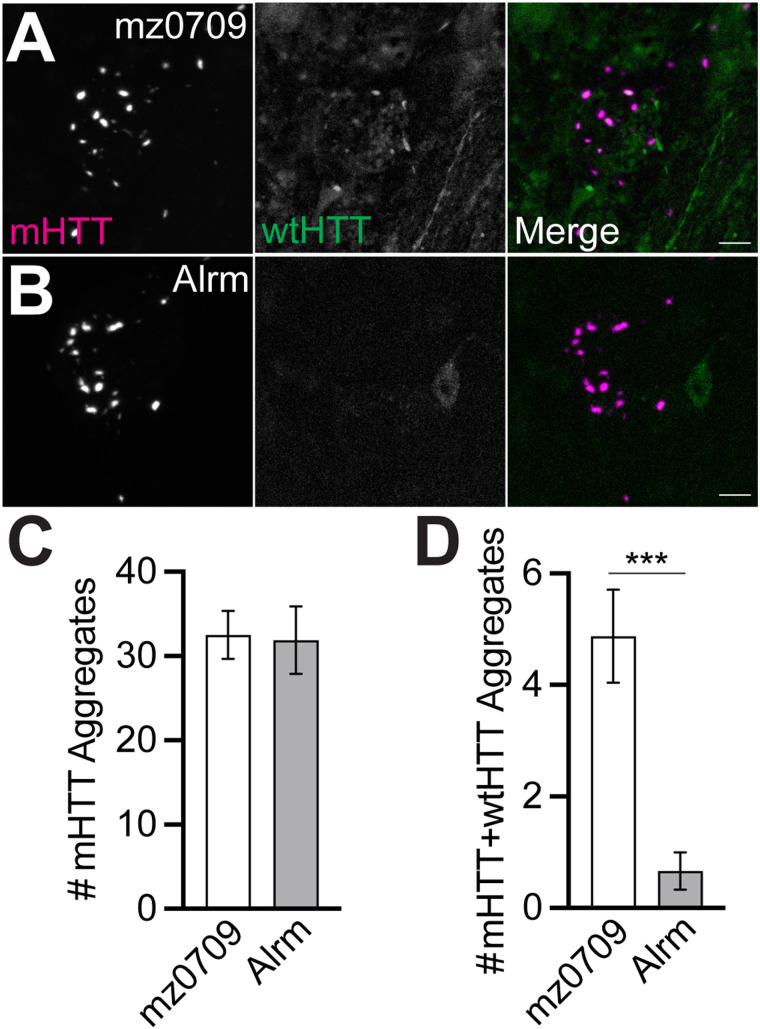
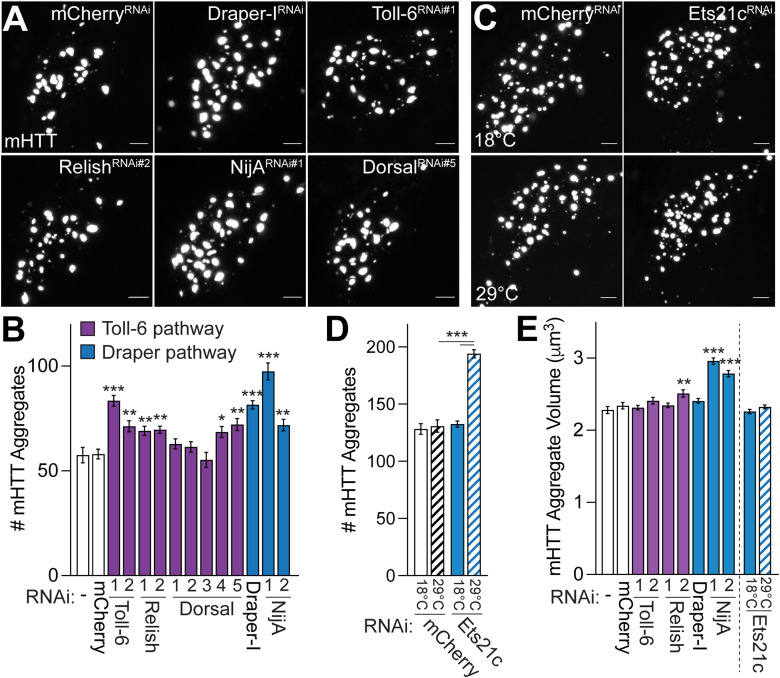
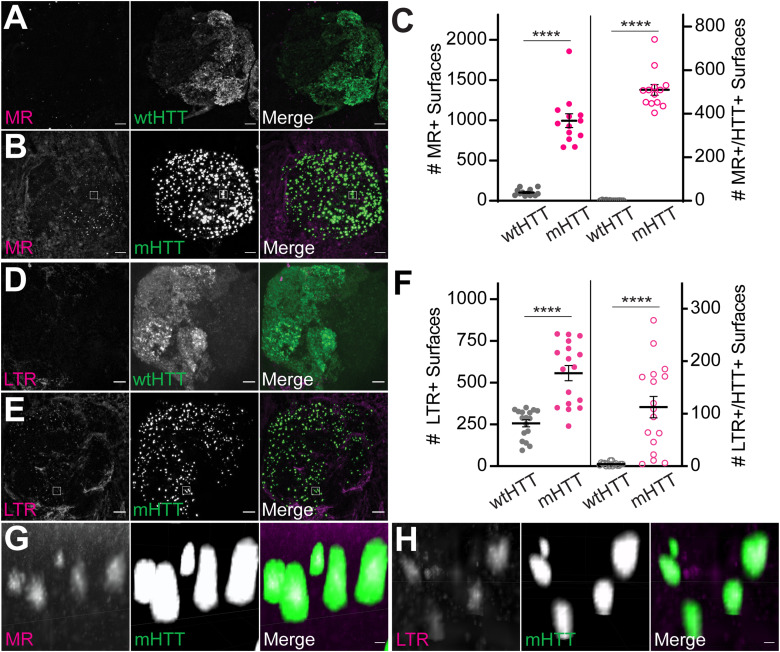
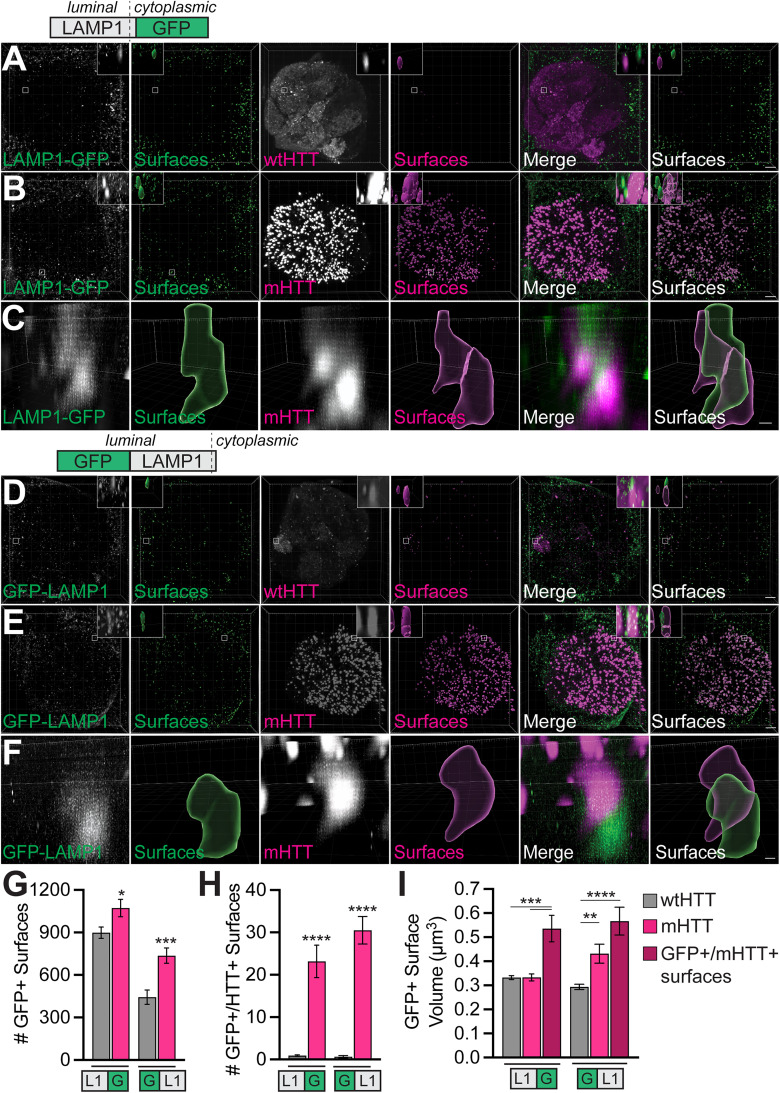
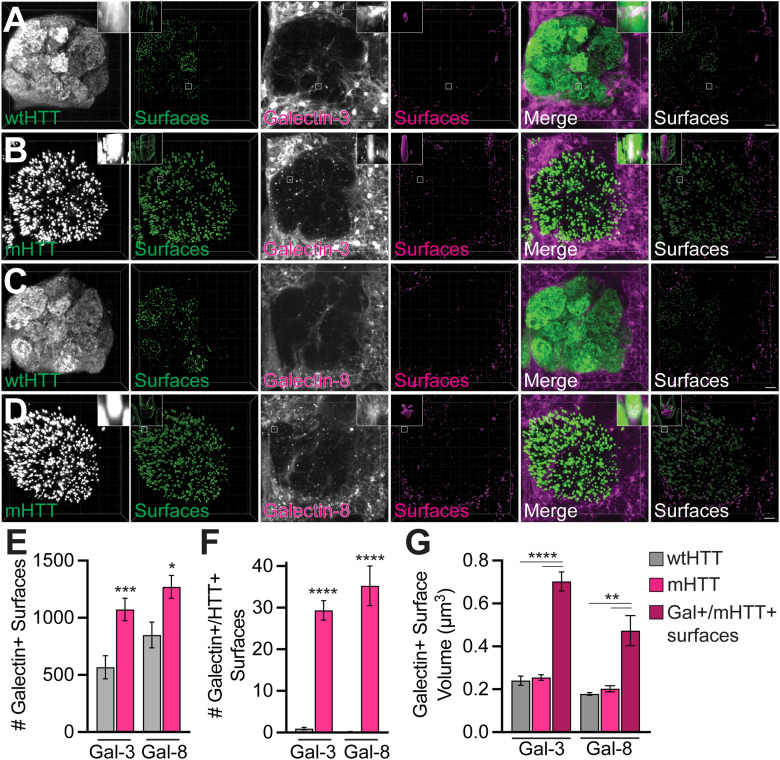
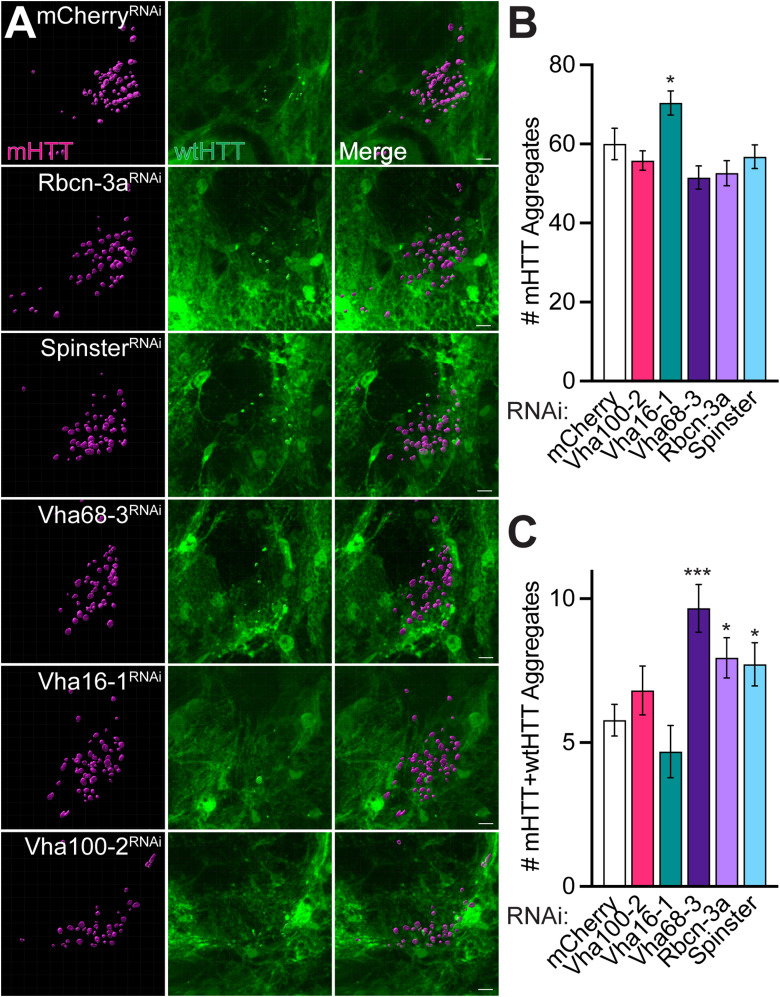
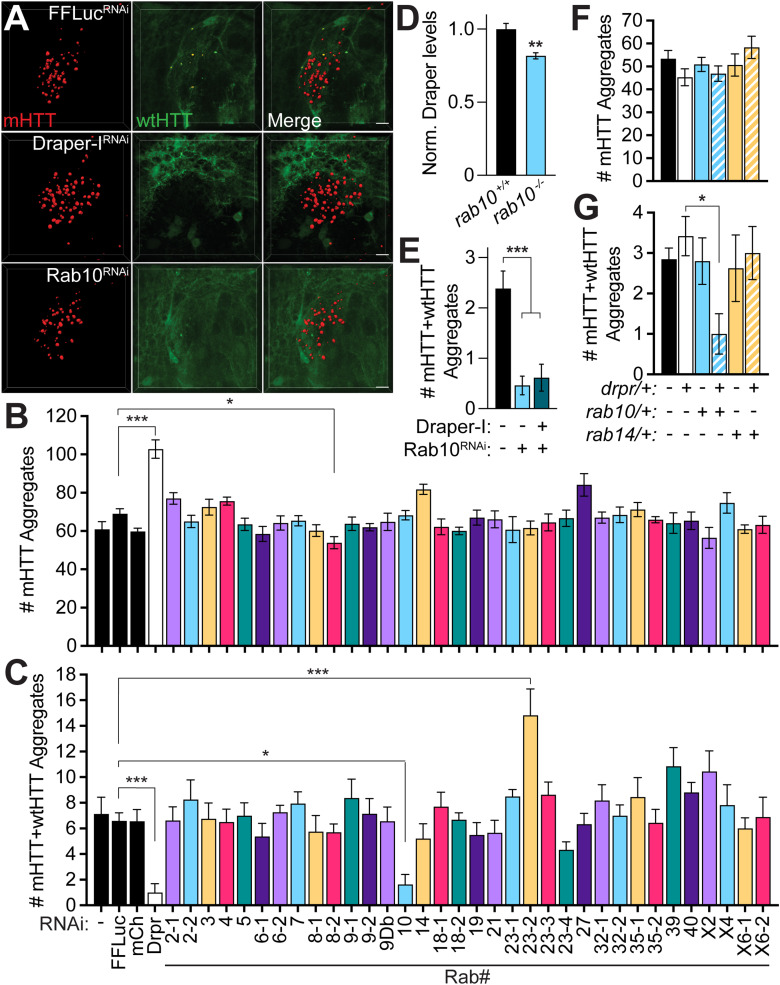
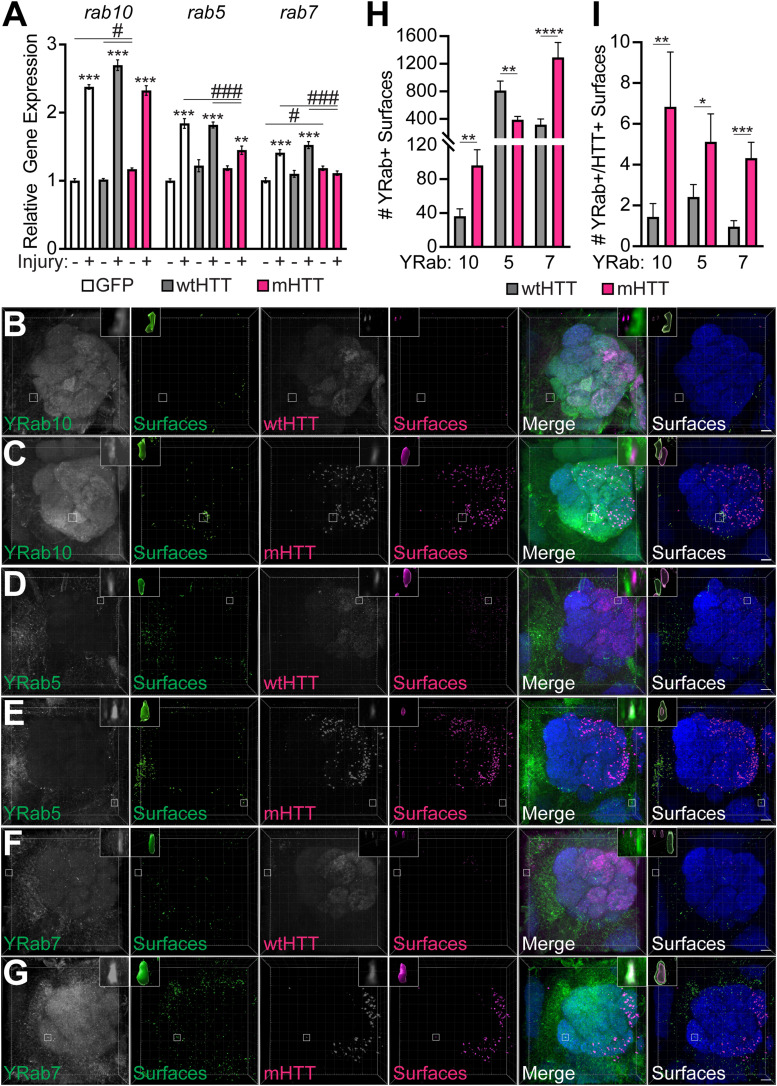
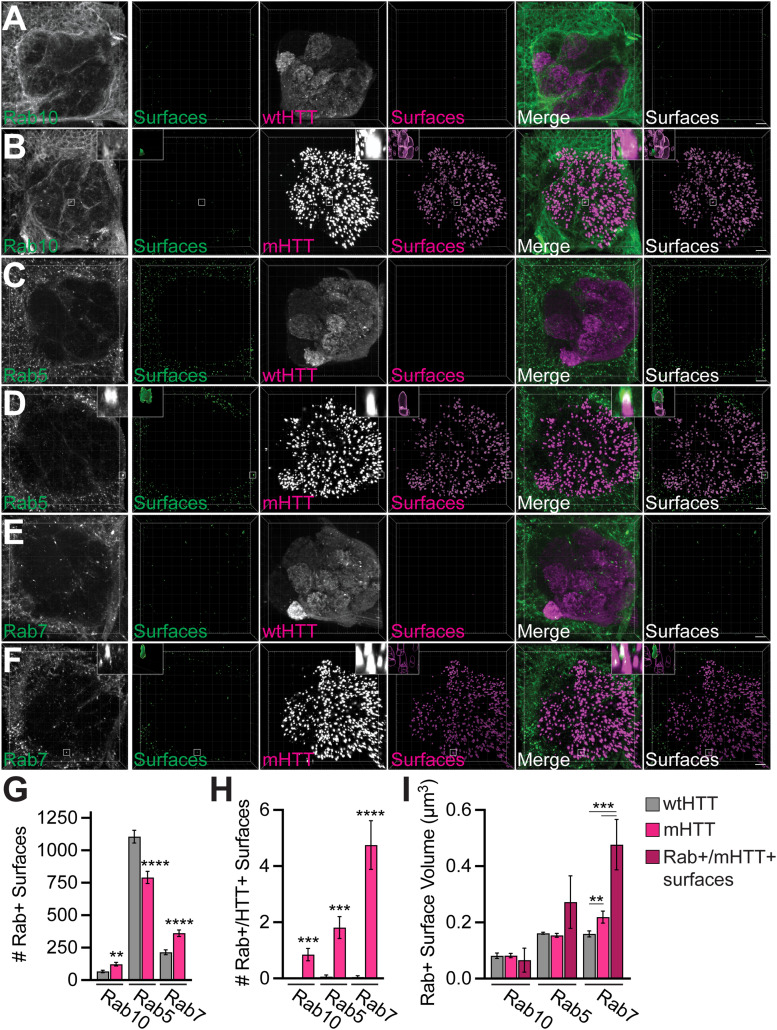
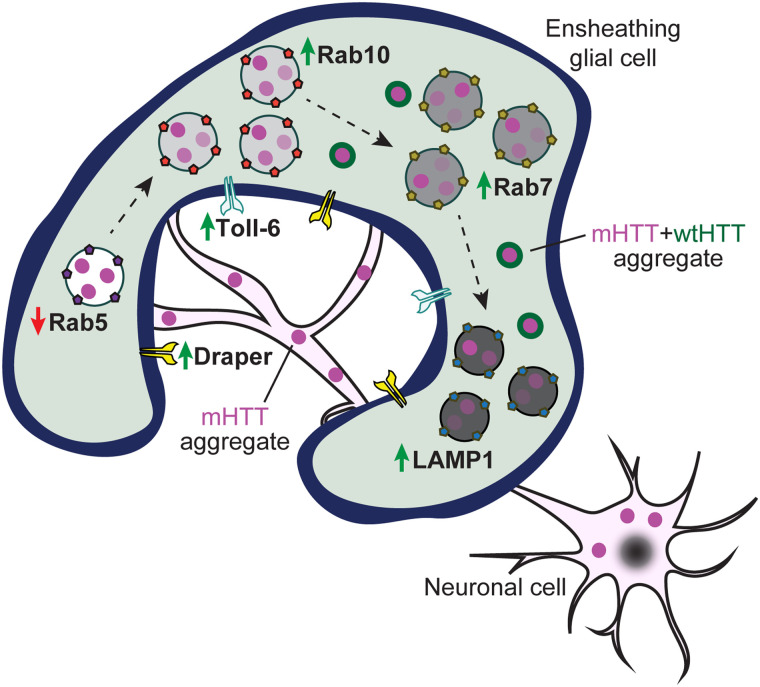
Protein misfolding, aggregation, and spread through the brain are primary drivers of neurodegenerative disease pathogenesis. Phagocytic glia are responsible for regulating the load of pathological proteins in the brain, but emerging evidence suggests that glia may also act as vectors for aggregate spread. Accumulation of protein aggregates could compromise the ability of glia to eliminate toxic materials from the brain by disrupting efficient degradation in the phagolysosomal system. A better understanding of phagocytic glial cell deficiencies in the disease state could help to identify novel therapeutic targets for multiple neurological disorders. Here, we report that mutant huntingtin (mHTT) aggregates impair glial responsiveness to injury and capacity to degrade neuronal debris in male and female adult Drosophila expressing the gene that causes Huntington's disease (HD). mHTT aggregate formation in neurons impairs engulfment and clearance of injured axons and causes accumulation of phagolysosomes in glia. Neuronal mHTT expression induces upregulation of key innate immunity and phagocytic genes, some of which were found to regulate mHTT aggregate burden in the brain. A forward genetic screen revealed Rab10 as a novel component of Draper-dependent phagocytosis that regulates mHTT aggregate transmission from neurons to glia. These data suggest that glial phagocytic defects enable engulfed mHTT aggregates to evade lysosomal degradation and acquire prion-like characteristics. Together, our findings uncover new mechanisms that enhance our understanding of the beneficial and harmful effects of phagocytic glia in HD and other neurodegenerative diseases.
Keywords: Rab; aggregate; glia; huntingtin; phagocytosis; prion-like.
Copyright © 2024 the authors.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

Update of
-
Impairment of the glial phagolysosomal system drives prion-like propagation in a Drosophila model of Huntington's disease.bioRxiv [Preprint]. 2024 Feb 5:2023.10.04.560952. doi: 10.1101/2023.10.04.560952. bioRxiv. 2024. Update in: J Neurosci. 2024 May 15;44(20):e1256232024. doi: 10.1523/JNEUROSCI.1256-23.2024 PMID: 38370619 Free PMC article. Updated. Preprint.
Similar articles
-
Impairment of the glial phagolysosomal system drives prion-like propagation in a Drosophila model of Huntington's disease.bioRxiv [Preprint]. 2024 Feb 5:2023.10.04.560952. doi: 10.1101/2023.10.04.560952. bioRxiv. 2024. Update in: J Neurosci. 2024 May 15;44(20):e1256232024. doi: 10.1523/JNEUROSCI.1256-23.2024 PMID: 38370619 Free PMC article. Updated. Preprint.
-
Phagocytic glia are obligatory intermediates in transmission of mutant huntingtin aggregates across neuronal synapses.Elife. 2020 May 28;9:e58499. doi: 10.7554/eLife.58499. Elife. 2020. PMID: 32463364 Free PMC article.
-
Downregulation of glial genes involved in synaptic function mitigates Huntington's disease pathogenesis.Elife. 2021 Apr 19;10:e64564. doi: 10.7554/eLife.64564. Elife. 2021. PMID: 33871358 Free PMC article.
-
Visualization of prion-like transfer in Huntington's disease models.Biochim Biophys Acta Mol Basis Dis. 2017 Mar;1863(3):793-800. doi: 10.1016/j.bbadis.2016.12.015. Epub 2016 Dec 29. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 28040507 Review.
-
Prion-like properties of the mutant huntingtin protein in living organisms: the evidence and the relevance.Mol Psychiatry. 2022 Jan;27(1):269-280. doi: 10.1038/s41380-021-01350-4. Mol Psychiatry. 2022. PMID: 34711942 Review.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases