

The cell biology of HIV-1 latency and rebound
- PMID: 38580979
- PMCID: PMC10996279
- DOI: 10.1186/s12977-024-00639-w
The cell biology of HIV-1 latency and rebound
Abstract
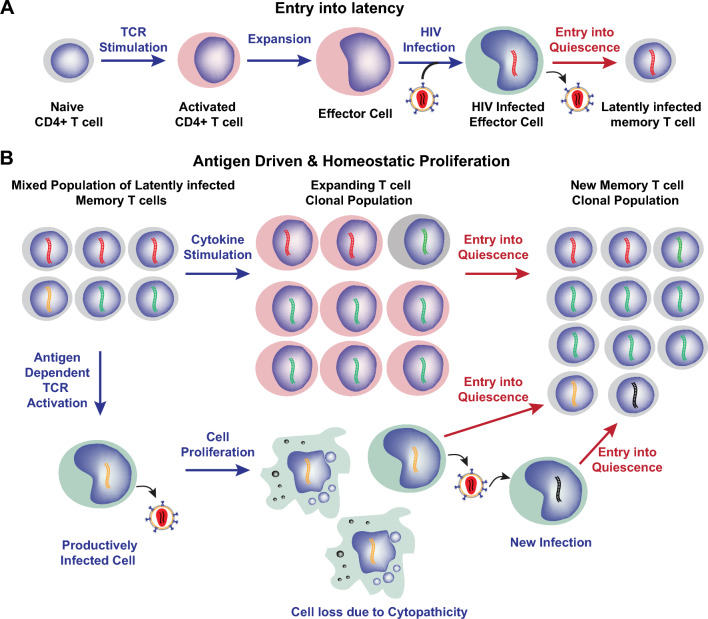
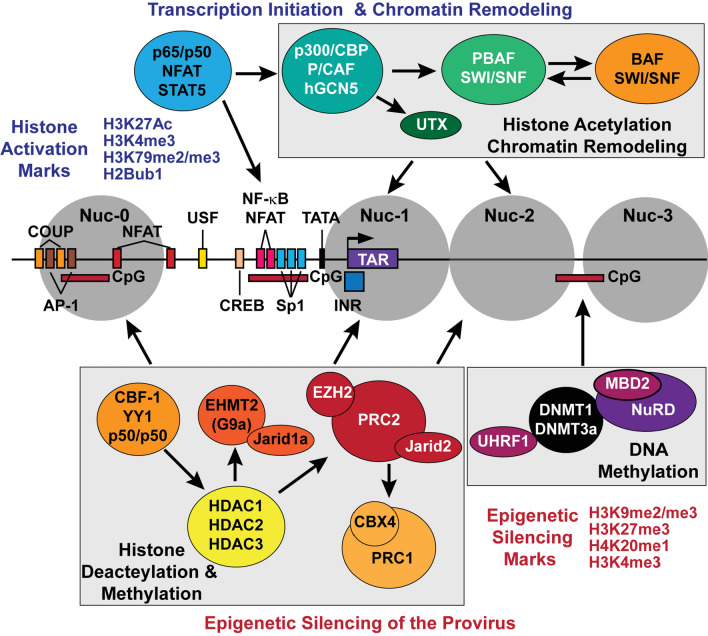
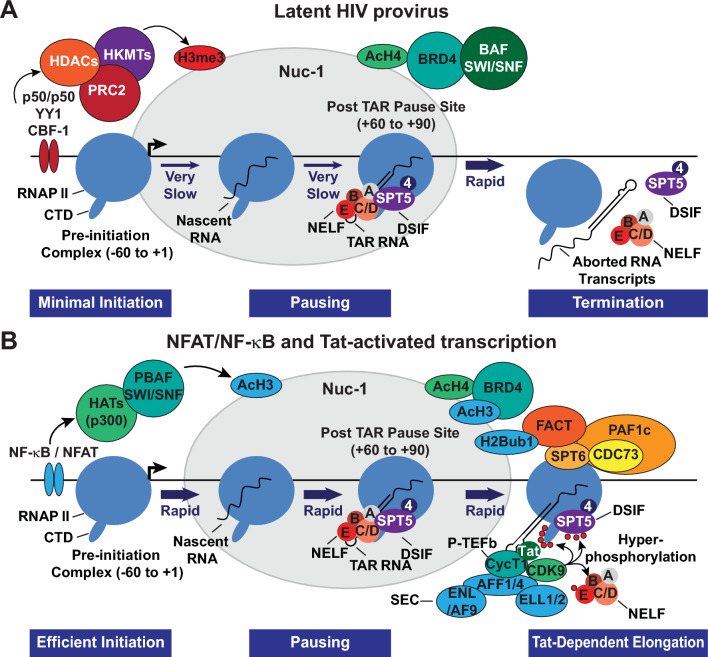
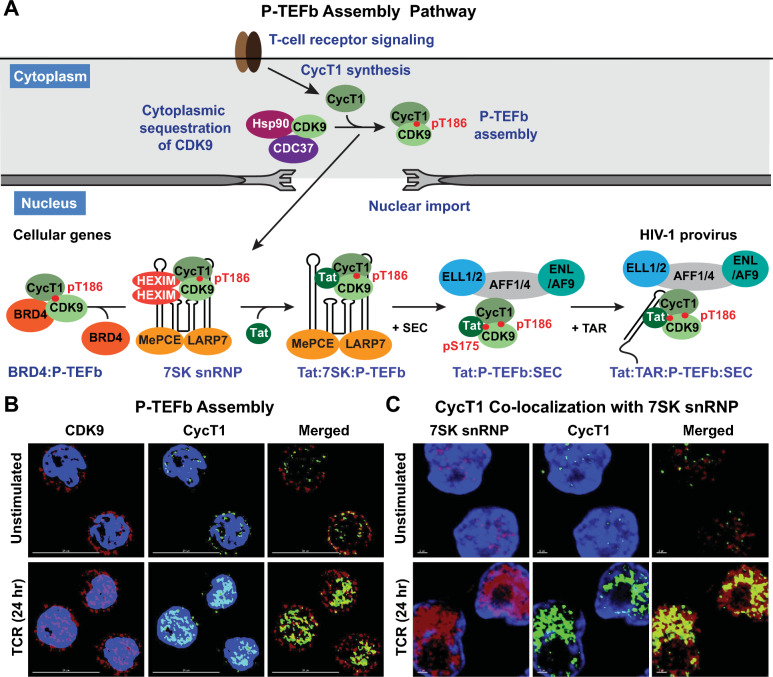
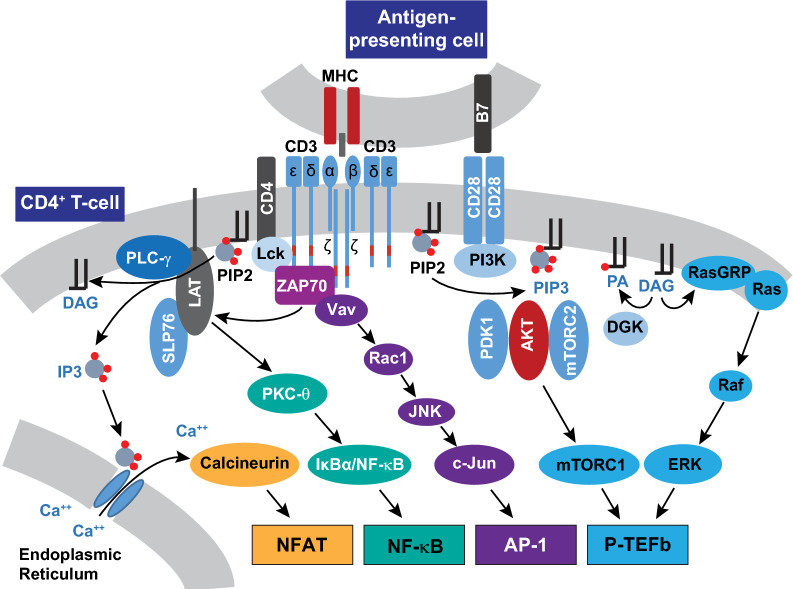
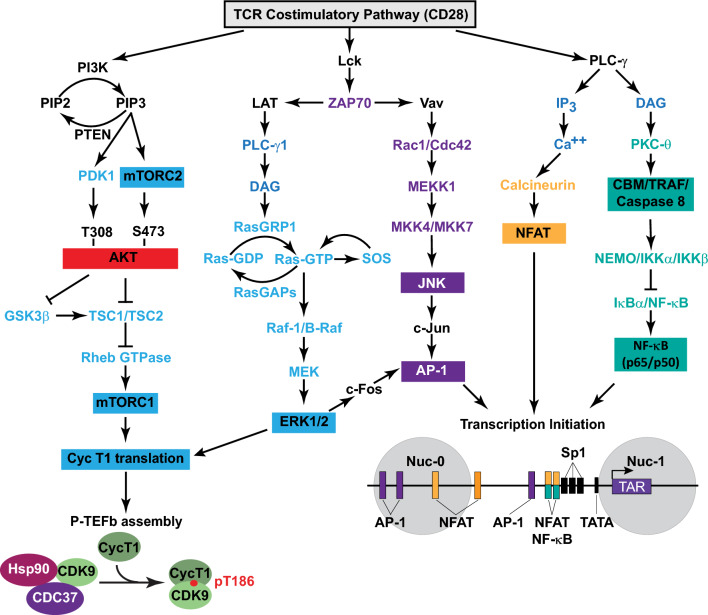
Transcriptionally latent forms of replication-competent proviruses, present primarily in a small subset of memory CD4+ T cells, pose the primary barrier to a cure for HIV-1 infection because they are the source of the viral rebound that almost inevitably follows the interruption of antiretroviral therapy. Over the last 30 years, many of the factors essential for initiating HIV-1 transcription have been identified in studies performed using transformed cell lines, such as the Jurkat T-cell model. However, as highlighted in this review, several poorly understood mechanisms still need to be elucidated, including the molecular basis for promoter-proximal pausing of the transcribing complex and the detailed mechanism of the delivery of P-TEFb from 7SK snRNP. Furthermore, the central paradox of HIV-1 transcription remains unsolved: how are the initial rounds of transcription achieved in the absence of Tat? A critical limitation of the transformed cell models is that they do not recapitulate the transitions between active effector cells and quiescent memory T cells. Therefore, investigation of the molecular mechanisms of HIV-1 latency reversal and LRA efficacy in a proper physiological context requires the utilization of primary cell models. Recent mechanistic studies of HIV-1 transcription using latently infected cells recovered from donors and ex vivo cellular models of viral latency have demonstrated that the primary blocks to HIV-1 transcription in memory CD4+ T cells are restrictive epigenetic features at the proviral promoter, the cytoplasmic sequestration of key transcription initiation factors such as NFAT and NF-κB, and the vanishingly low expression of the cellular transcription elongation factor P-TEFb. One of the foremost schemes to eliminate the residual reservoir is to deliberately reactivate latent HIV-1 proviruses to enable clearance of persisting latently infected cells-the "Shock and Kill" strategy. For "Shock and Kill" to become efficient, effective, non-toxic latency-reversing agents (LRAs) must be discovered. Since multiple restrictions limit viral reactivation in primary cells, understanding the T-cell signaling mechanisms that are essential for stimulating P-TEFb biogenesis, initiation factor activation, and reversing the proviral epigenetic restrictions have become a prerequisite for the development of more effective LRAs.
Keywords: 7SK snRNP; Epigenetic silencing; HIV-1 Tat; HIV-1 latency; HIV-1 reservoir; Latency reversal; P-TEFb; T-cell receptor signaling; Transcription elongation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
New insights into transcription elongation control of HIV-1 latency and rebound.Trends Immunol. 2023 Jan;44(1):60-71. doi: 10.1016/j.it.2022.11.003. Epub 2022 Dec 8. Trends Immunol. 2023. PMID: 36503686 Review.
-
The Molecular Basis for Human Immunodeficiency Virus Latency.Annu Rev Virol. 2017 Sep 29;4(1):261-285. doi: 10.1146/annurev-virology-101416-041646. Epub 2017 Jul 17. Annu Rev Virol. 2017. PMID: 28715973 Review.
-
Alternate NF-κB-Independent Signaling Reactivation of Latent HIV-1 Provirus.J Virol. 2019 Aug 28;93(18):e00495-19. doi: 10.1128/JVI.00495-19. Print 2019 Sep 15. J Virol. 2019. PMID: 31243131 Free PMC article.
-
T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway.J Mol Biol. 2011 Jul 29;410(5):896-916. doi: 10.1016/j.jmb.2011.03.054. J Mol Biol. 2011. PMID: 21763495 Free PMC article.
-
Thiostrepton Reactivates Latent HIV-1 through the p-TEFb and NF-κB Pathways Mediated by Heat Shock Response.Antimicrob Agents Chemother. 2020 Apr 21;64(5):e02328-19. doi: 10.1128/AAC.02328-19. Print 2020 Apr 21. Antimicrob Agents Chemother. 2020. PMID: 32094131 Free PMC article.
Cited by
-
Protocol for intracellular immunofluorescence measurements of latent HIV reactivation in a primary CD4+ T cell model.STAR Protoc. 2024 Dec 20;5(4):103398. doi: 10.1016/j.xpro.2024.103398. Epub 2024 Oct 18. STAR Protoc. 2024. PMID: 39425934 Free PMC article.
-
Integrator complex subunit 12 knockout overcomes a transcriptional block to HIV latency reversal.bioRxiv [Preprint]. 2025 Feb 19:2024.08.30.610517. doi: 10.1101/2024.08.30.610517. bioRxiv. 2025. PMID: 39257755 Free PMC article. Preprint.
-
HIV Persistence, Latency, and Cure Approaches: Where Are We Now?Viruses. 2024 Jul 19;16(7):1163. doi: 10.3390/v16071163. Viruses. 2024. PMID: 39066325 Free PMC article. Review.
References
-
- Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9(3):e1003211. doi: 10.1371/journal.ppat.1003211. - DOI - PMC - PubMed
-
- Hocqueloux L, Prazuck T, Avettand-Fenoel V, Lafeuillade A, Cardon B, Viard JP, Rouzioux C. Long-term immunovirologic control following antiretroviral therapy interruption in patients treated at the time of primary HIV-1 infection. AIDS. 2010;24(10):1598–1601. doi: 10.1097/qad.0b013e32833b61ba. - DOI - PubMed
-
- Davey RT, Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A. 1999;96(26):15109–15114. doi: 10.1073/pnas.96.26.15109. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
