

PDCD4 restricts PRRSV replication in an eIF4A-dependent manner and is antagonized by the viral nonstructural protein 9
- PMID: 38557170
- PMCID: PMC11092367
- DOI: 10.1128/jvi.00060-24
PDCD4 restricts PRRSV replication in an eIF4A-dependent manner and is antagonized by the viral nonstructural protein 9
Abstract
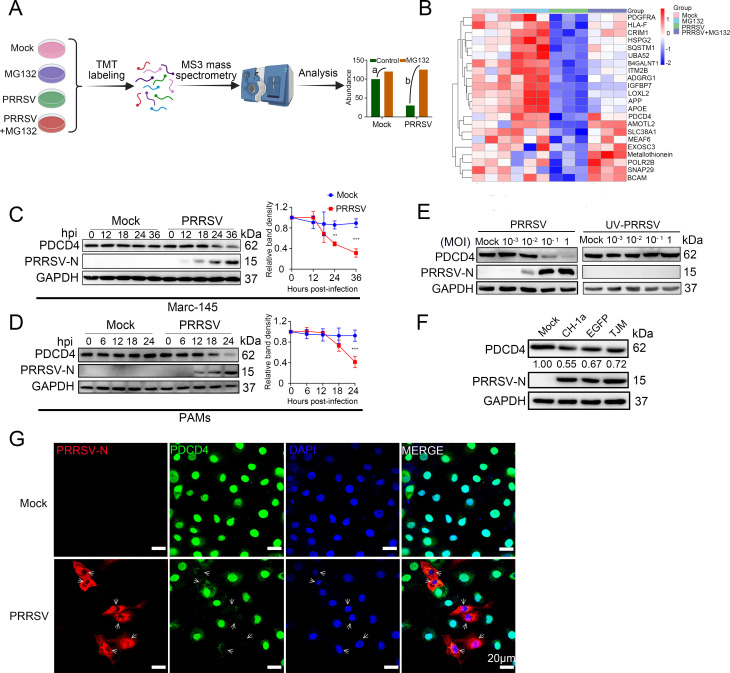
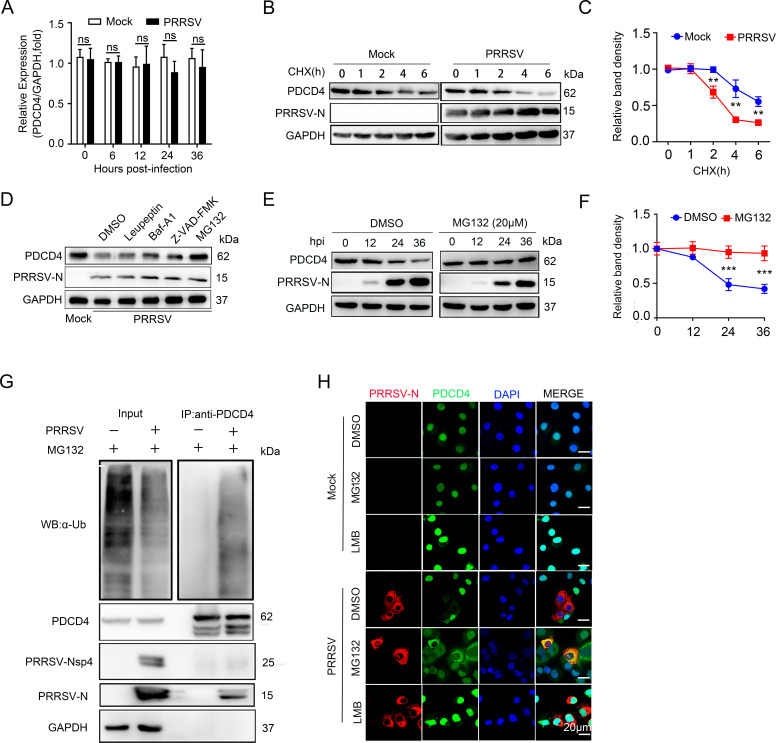
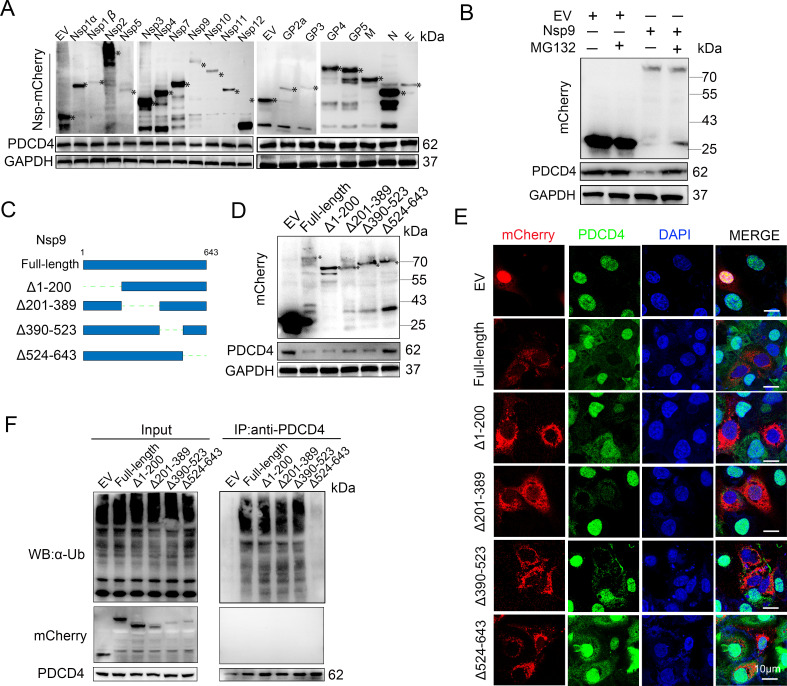
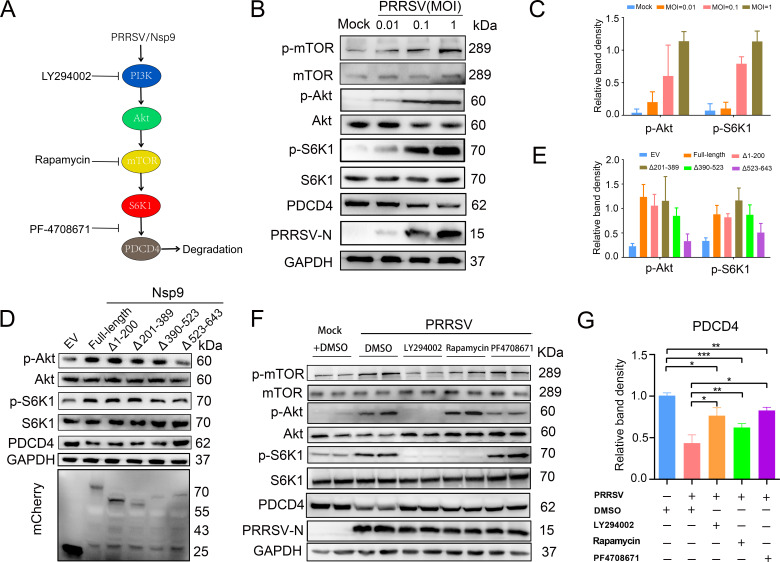
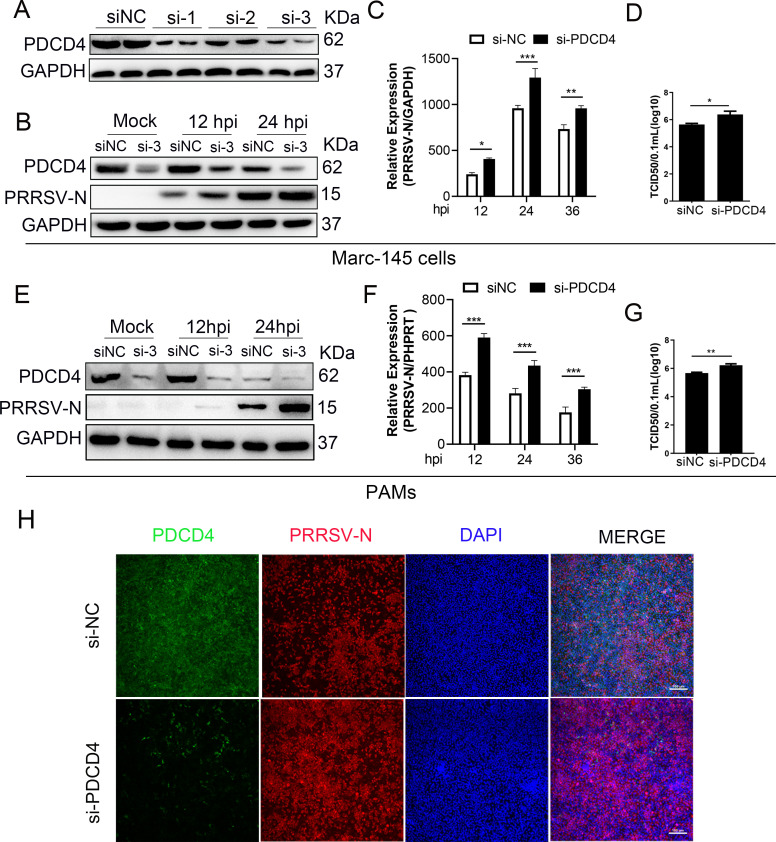
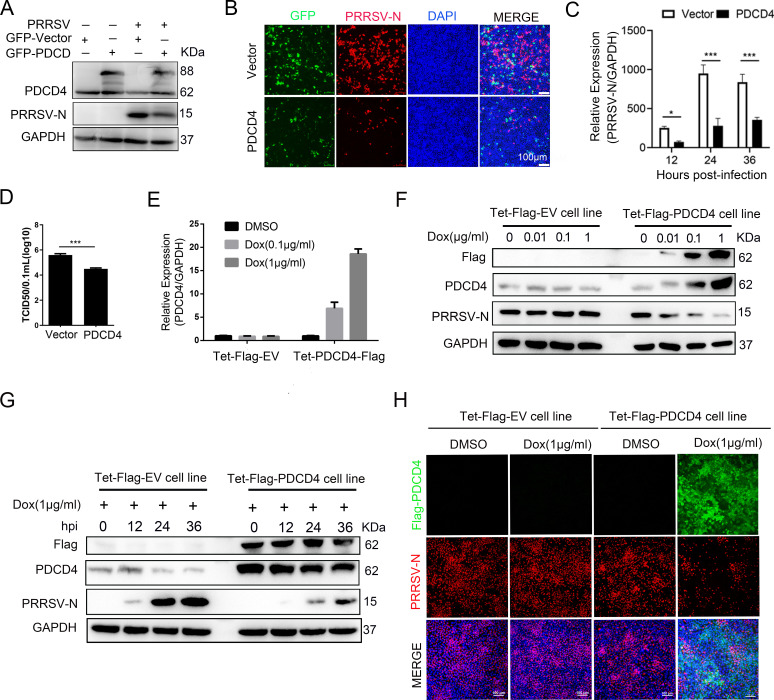
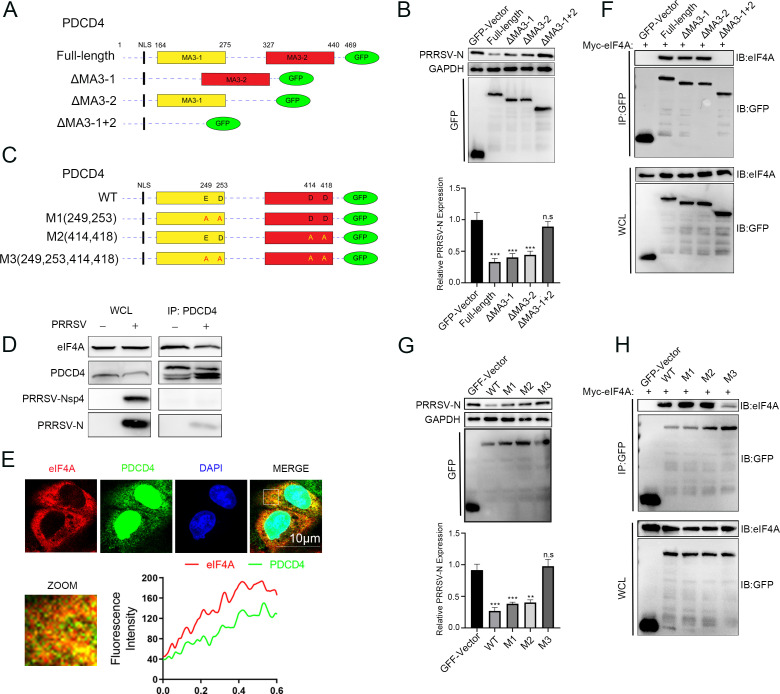
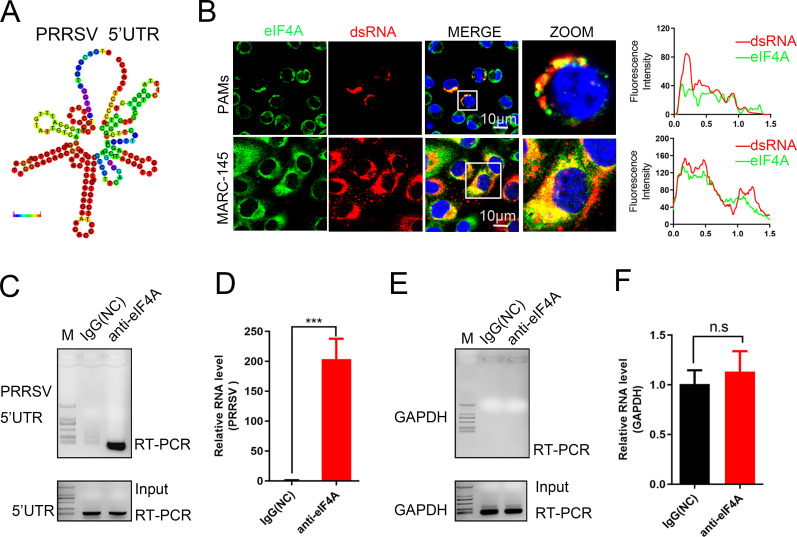
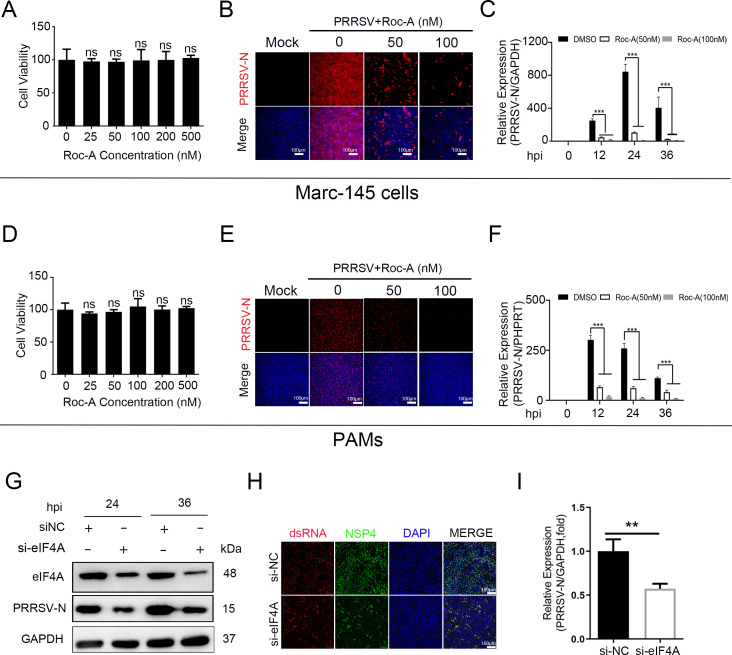
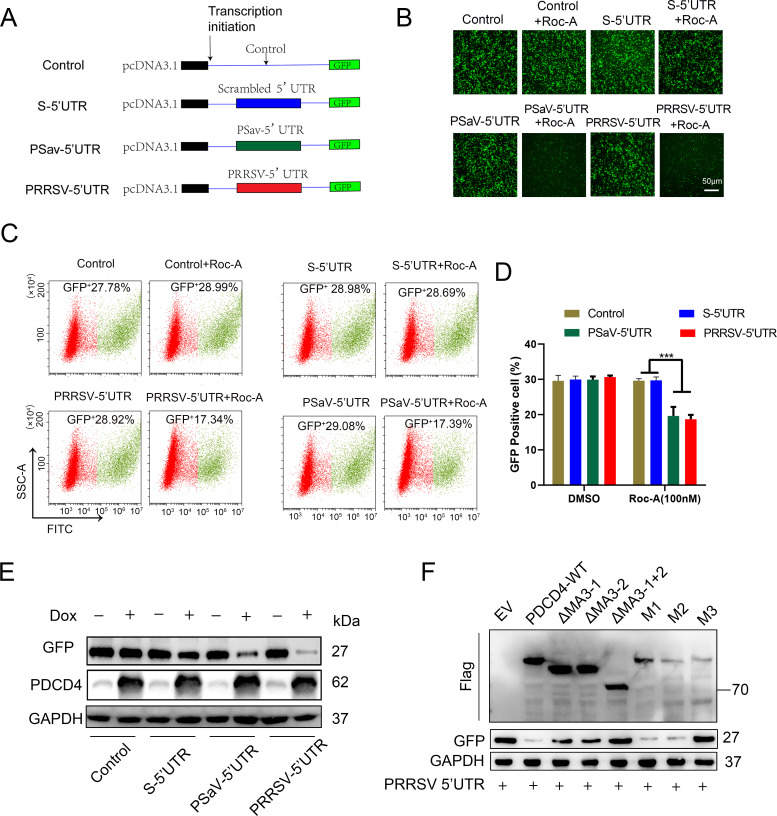
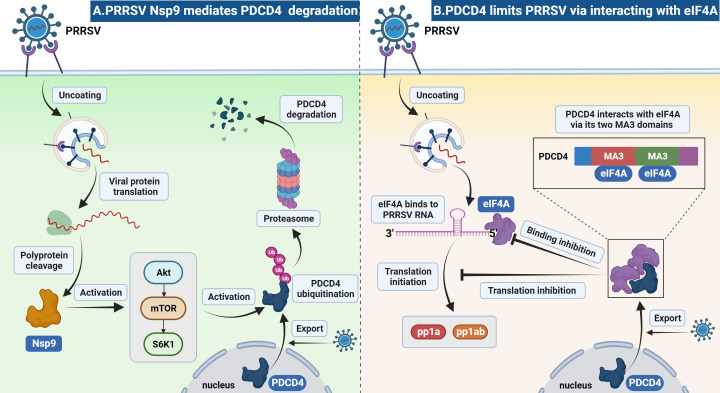
As obligate parasites, viruses have evolved multiple strategies to evade the host immune defense. Manipulation of the host proteasome system to degrade specific detrimental factors is a common viral countermeasure. To identify host proteins targeted for proteasomal degradation by porcine reproductive and respiratory syndrome virus (PRRSV), we conducted a quantitative proteomics screen of PRRSV-infected Marc-145 cells under the treatment with proteasome inhibitor MG132. The data revealed that the expression levels of programmed cell death 4 (PDCD4) were strongly downregulated by PRRSV and significantly rescued by MG132. Further investigation confirmed that PRRSV infection induced the translocation of PDCD4 from the nucleus to the cytoplasm, and the viral nonstructural protein 9 (Nsp9) promoted PDCD4 proteasomal degradation in the cytoplasm by activating the Akt-mTOR-S6K1 pathway. The C-terminal domain of Nsp9 was responsible for PDCD4 degradation. As for the role of PDCD4 during PRRSV infection, we demonstrated that PDCD4 knockdown favored viral replication, while its overexpression significantly attenuated replication, suggesting that PDCD4 acts as a restriction factor for PRRSV. Mechanistically, we discovered eukaryotic translation initiation factor 4A (eIF4A) was required for PRRSV. PDCD4 interacted with eIF4A through four sites (E249, D253, D414, and D418) within its two MA3 domains, disrupting eIF4A-mediated translation initiation in the 5'-untranslated region of PRRSV, thereby inhibiting PRRSV infection. Together, our study reveals the antiviral function of PDCD4 and the viral strategy to antagonize PDCD4. These results will contribute to our understanding of the immune evasion strategies employed by PRRSV and offer valuable insights for developing new antiviral targets.IMPORTANCEPorcine reproductive and respiratory syndrome virus (PRRSV) infection results in major economic losses in the global swine industry and is difficult to control effectively. Here, using a quantitative proteomics screen, we identified programmed cell death 4 (PDCD4) as a host protein targeted for proteasomal degradation by PRRSV. We demonstrated that PDCD4 restricts PRRSV replication by interacting with eukaryotic translation initiation factor 4A, which is required for translation initiation in the viral 5'-untranslated region. Additionally, four sites within two MA3 domains of PDCD4 are identified to be responsible for its antiviral function. Conversely, PRRSV nonstructural protein 9 promotes PDCD4 proteasomal degradation in the cytoplasm by activating the Akt-mTOR-S6K1 pathway, thus weakening the anti-PRRSV function. Our work unveils PDCD4 as a previously unrecognized host restriction factor for PRRSV and reveals that PRRSV develops countermeasures to overcome PDCD4. This will provide new insights into virus-host interactions and the development of new antiviral targets.
Keywords: Nsp9; PDCD4; PRRSV; eIF4A; host restriction factor; virus-host interactions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Porcine Reproductive and Respiratory Syndrome Virus Infection Induces both eIF2α Phosphorylation-Dependent and -Independent Host Translation Shutoff.J Virol. 2018 Jul 31;92(16):e00600-18. doi: 10.1128/JVI.00600-18. Print 2018 Aug 15. J Virol. 2018. PMID: 29899101 Free PMC article.
-
Porcine Reproductive and Respiratory Syndrome Virus Antagonizes JAK/STAT3 Signaling via nsp5, Which Induces STAT3 Degradation.J Virol. 2017 Jan 18;91(3):e02087-16. doi: 10.1128/JVI.02087-16. Print 2017 Feb 1. J Virol. 2017. PMID: 27881658 Free PMC article.
-
The interaction of nonstructural protein 9 with retinoblastoma protein benefits the replication of genotype 2 porcine reproductive and respiratory syndrome virus in vitro.Virology. 2014 Sep;464-465:432-440. doi: 10.1016/j.virol.2014.07.036. Epub 2014 Aug 21. Virology. 2014. PMID: 25146601 Free PMC article.
-
Role of phosphatidylinositol-3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) signalling pathways in porcine reproductive and respiratory syndrome virus (PRRSV) replication.Virus Res. 2014 Dec 19;194:138-44. doi: 10.1016/j.virusres.2014.09.017. Epub 2014 Oct 7. Virus Res. 2014. PMID: 25304692 Review.
-
Host combats porcine reproductive and respiratory syndrome virus infection at non-coding RNAs level.Virulence. 2024 Dec;15(1):2416551. doi: 10.1080/21505594.2024.2416551. Epub 2024 Oct 18. Virulence. 2024. PMID: 39403796 Free PMC article. Review.
References
-
- Neumann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH, Green AL, Zimmerman JJ. 2005. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc 227:385–392. doi:10.2460/javma.2005.227.385 - DOI - PubMed
-
- Harding JCS, Ladinig A, Novakovic P, Detmer SE, Wilkinson JM, Yang T, Lunney JK, Plastow GS. 2017. Novel insights into host responses and reproductive pathophysiology of porcine reproductive and respiratory syndrome caused by PRRSV-2. Vet Microbiol 209:114–123. doi:10.1016/j.vetmic.2017.02.019 - DOI - PubMed
MeSH terms
Grants and funding
- 2023YFD1801500/MOST | National Key Research and Development Program of China (NKPs)
- 32122082, 32072695/MOST | National Natural Science Foundation of China (NSFC)
- 32302722/MOST | National Natural Science Foundation of China (NSFC)
- 2023B03J0947/Science and Technology Planning Project of Guangzhou
- NG2022003/Laboratory of Lingnan Modern Agriculture Project
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
