

A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche
- PMID: 38509359
- PMCID: PMC11006615
- DOI: 10.1038/s41586-024-07182-w
A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche
Abstract
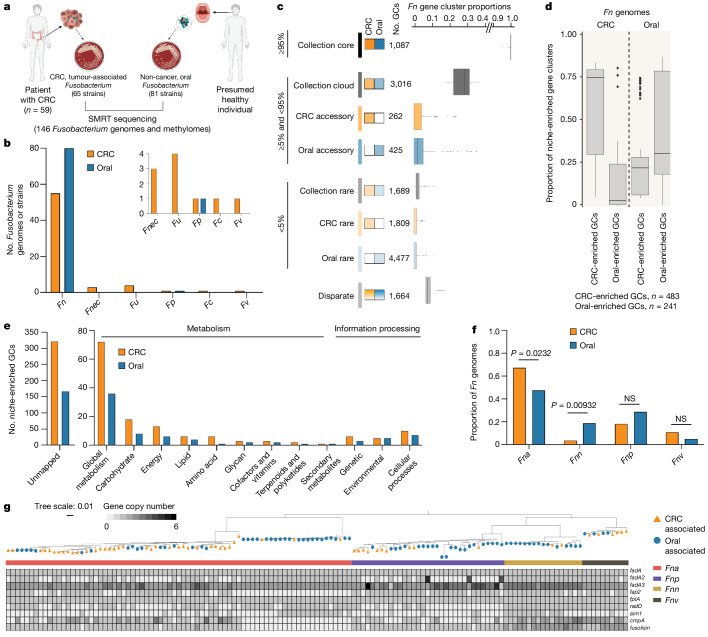
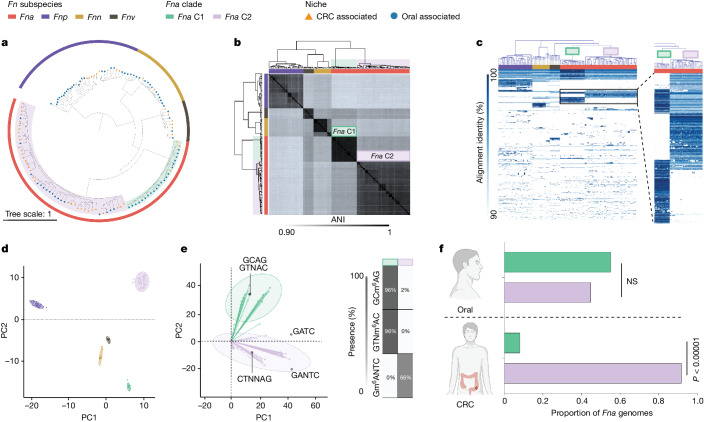
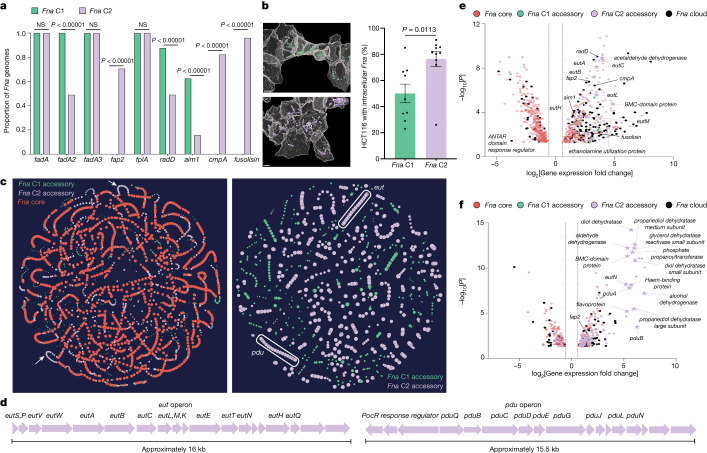
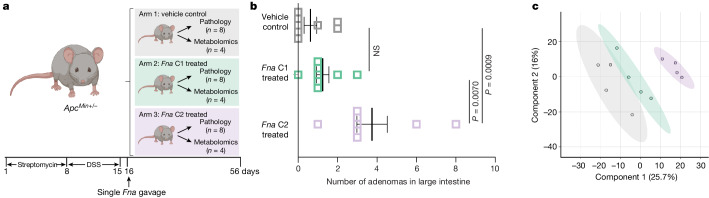
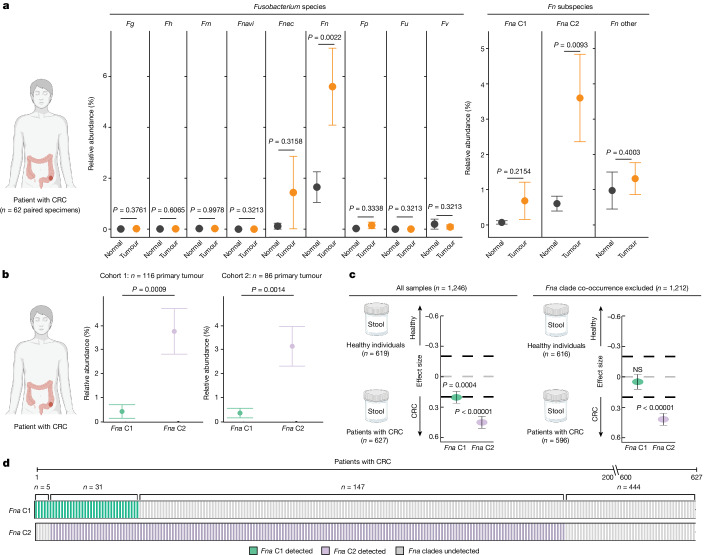
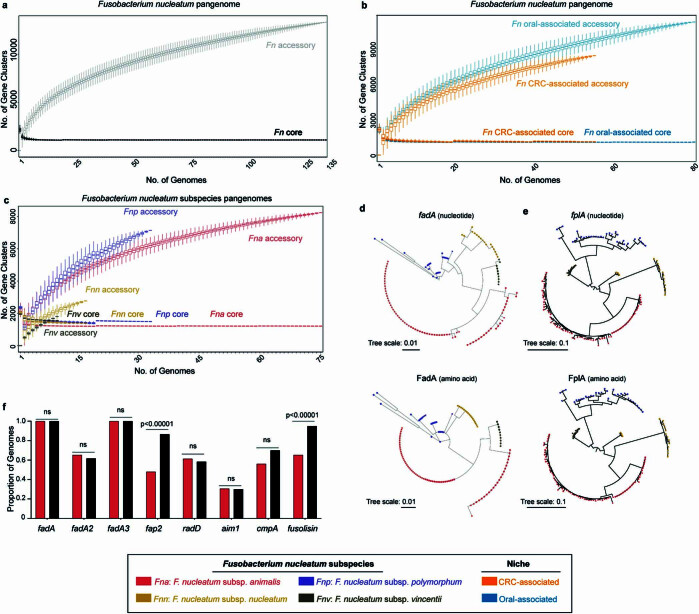
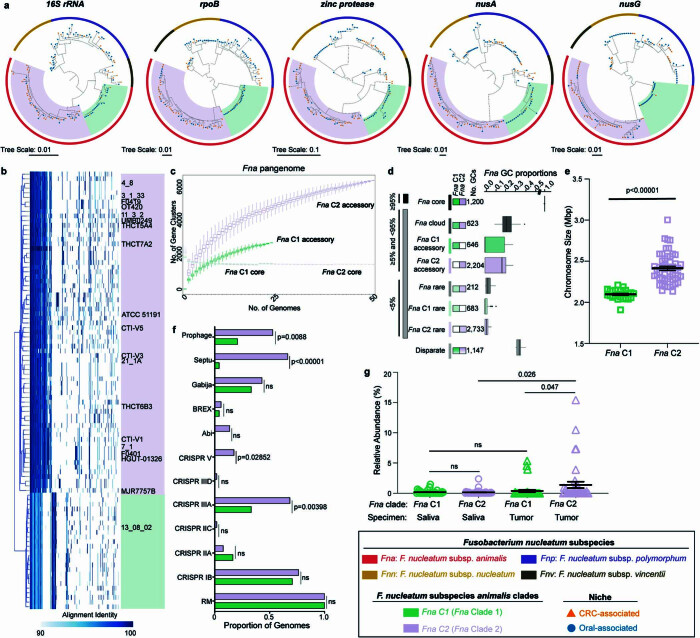
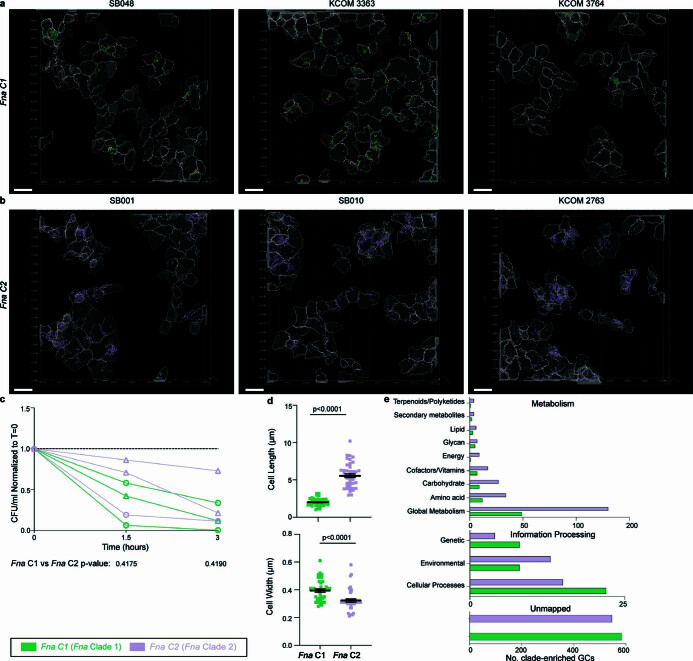
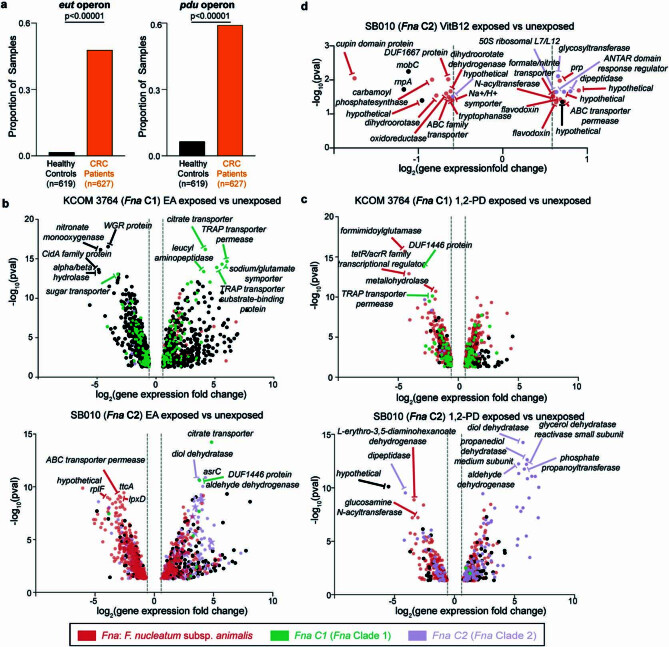
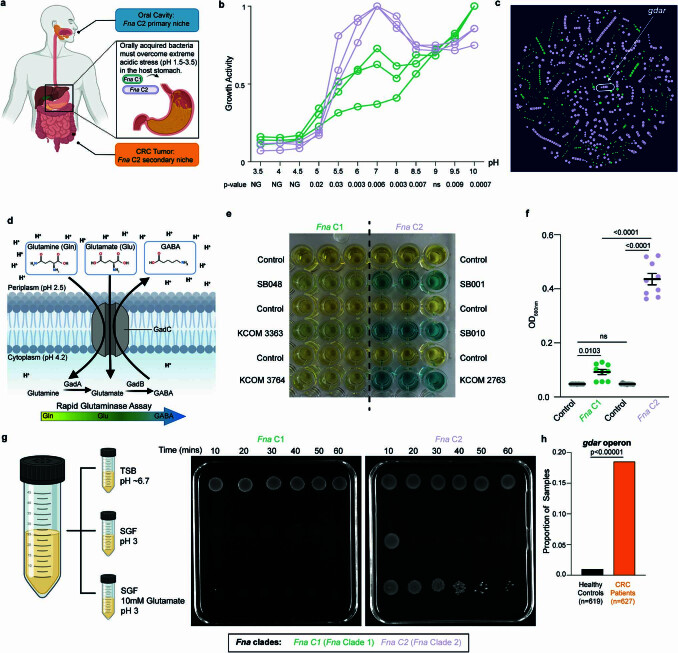
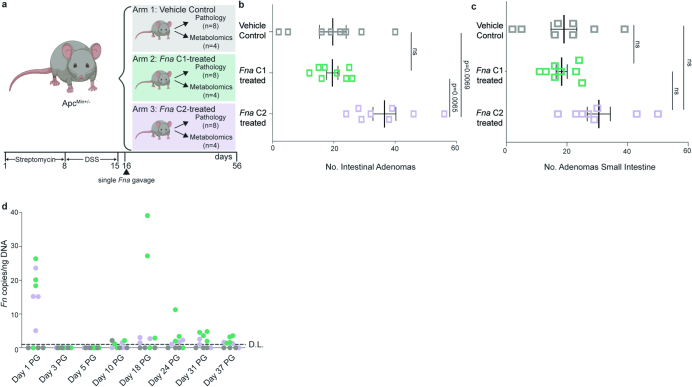
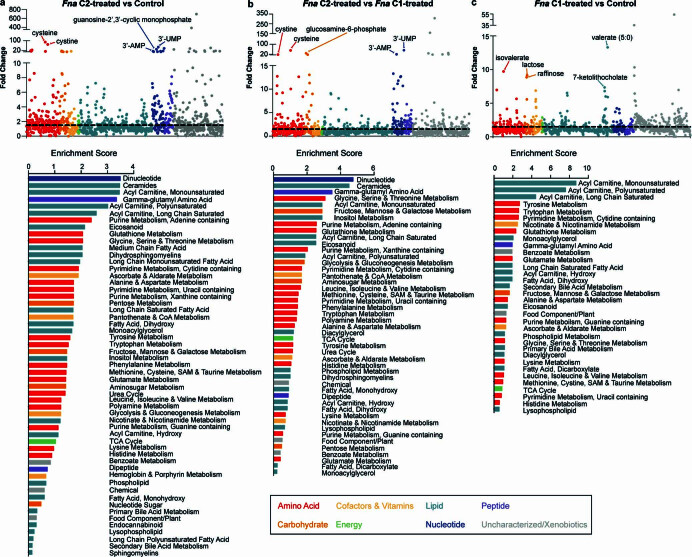
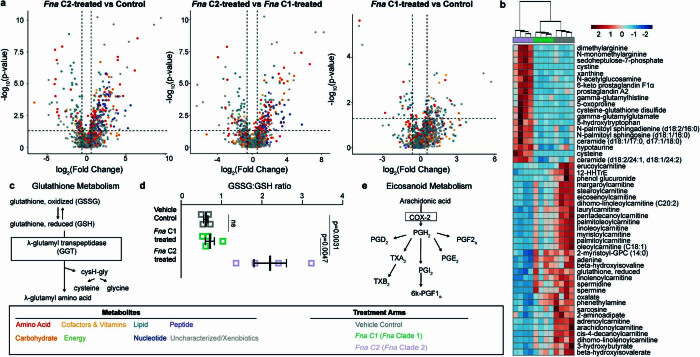
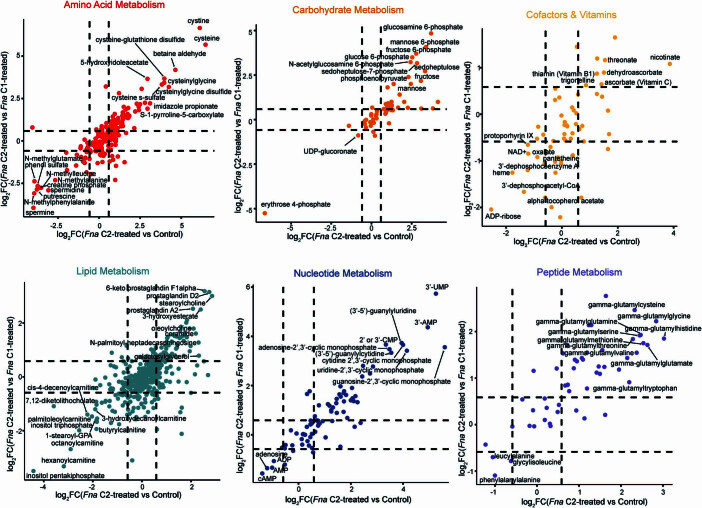
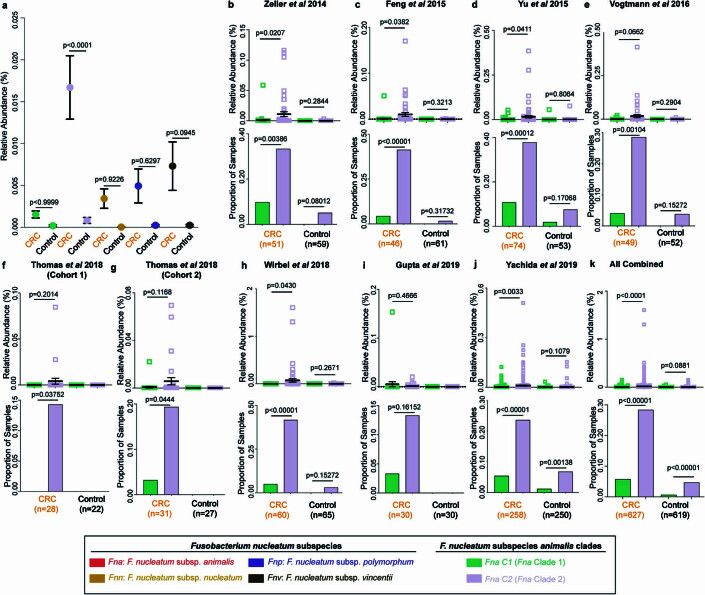
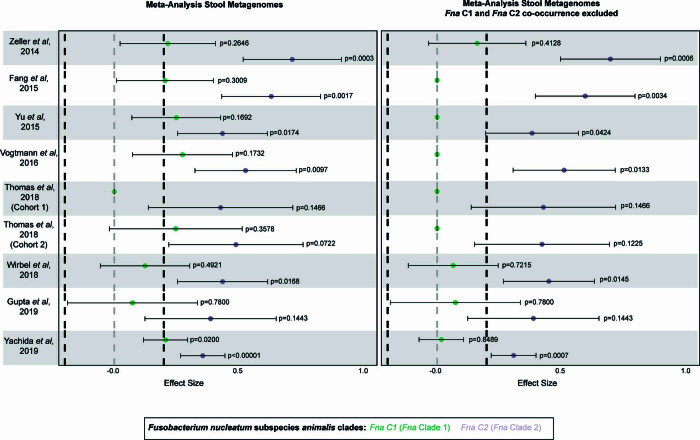
Fusobacterium nucleatum (Fn), a bacterium present in the human oral cavity and rarely found in the lower gastrointestinal tract of healthy individuals1, is enriched in human colorectal cancer (CRC) tumours2-5. High intratumoural Fn loads are associated with recurrence, metastases and poorer patient prognosis5-8. Here, to delineate Fn genetic factors facilitating tumour colonization, we generated closed genomes for 135 Fn strains; 80 oral strains from individuals without cancer and 55 unique cancer strains cultured from tumours from 51 patients with CRC. Pangenomic analyses identified 483 CRC-enriched genetic factors. Tumour-isolated strains predominantly belong to Fn subspecies animalis (Fna). However, genomic analyses reveal that Fna, considered a single subspecies, is instead composed of two distinct clades (Fna C1 and Fna C2). Of these, only Fna C2 dominates the CRC tumour niche. Inter-Fna analyses identified 195 Fna C2-associated genetic factors consistent with increased metabolic potential and colonization of the gastrointestinal tract. In support of this, Fna C2-treated mice had an increased number of intestinal adenomas and altered metabolites. Microbiome analysis of human tumour tissue from 116 patients with CRC demonstrated Fna C2 enrichment. Comparison of 62 paired specimens showed that only Fna C2 is tumour enriched compared to normal adjacent tissue. This was further supported by metagenomic analysis of stool samples from 627 patients with CRC and 619 healthy individuals. Collectively, our results identify the Fna clade bifurcation, show that specifically Fna C2 drives the reported Fn enrichment in human CRC and reveal the genetic underpinnings of pathoadaptation of Fna C2 to the CRC niche.
© 2024. The Author(s).
Conflict of interest statement
S.B. has consulted for GlaxoSmithKline and BiomX. C.D.J. has consulted for Series Therapeutics and Azitra. S.B. is an inventor on US patent application no. PCT/US2018/042966, submitted by the Broad Institute and Dana-Farber Cancer Institute, which covers the targeting of
Figures
Similar articles
-
Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome.Eur J Clin Microbiol Infect Dis. 2014 Aug;33(8):1381-90. doi: 10.1007/s10096-014-2081-3. Epub 2014 Mar 6. Eur J Clin Microbiol Infect Dis. 2014. PMID: 24599709
-
Aspirin Modulation of the Colorectal Cancer-Associated Microbe Fusobacterium nucleatum.mBio. 2021 Apr 6;12(2):e00547-21. doi: 10.1128/mBio.00547-21. mBio. 2021. PMID: 33824205 Free PMC article.
-
Highly sensitive stool DNA testing of Fusobacterium nucleatum as a marker for detection of colorectal tumours in a Japanese population.Ann Clin Biochem. 2017 Jan;54(1):86-91. doi: 10.1177/0004563216643970. Epub 2016 Sep 28. Ann Clin Biochem. 2017. PMID: 27126270
-
Fusobacterium nucleatum and colorectal cancer: A mechanistic overview.J Cell Physiol. 2019 Mar;234(3):2337-2344. doi: 10.1002/jcp.27250. Epub 2018 Sep 7. J Cell Physiol. 2019. PMID: 30191984 Review.
-
[Advances on the treatment of Fusobacterium nucleatum-promoted colorectal cancers using nanomaterials].Sheng Wu Gong Cheng Xue Bao. 2023 Sep 25;39(9):3670-3680. doi: 10.13345/j.cjb.220811. Sheng Wu Gong Cheng Xue Bao. 2023. PMID: 37805845 Review. Chinese.
Cited by
-
Perspectives on the future of host-microbe biology from the Council on Microbial Sciences of the American Society for Microbiology.mSphere. 2024 Jul 30;9(7):e0025624. doi: 10.1128/msphere.00256-24. Epub 2024 Jun 26. mSphere. 2024. PMID: 38920371 Free PMC article. Review.
-
Unravelling the role of intratumoral bacteria in digestive system cancers: current insights and future perspectives.J Transl Med. 2024 Jun 7;22(1):545. doi: 10.1186/s12967-024-05320-6. J Transl Med. 2024. PMID: 38849871 Free PMC article. Review.
-
Gut Microbiota Signatures in Colorectal Cancer as a Potential Diagnostic Biomarker in the Future: A Systematic Review.Int J Mol Sci. 2024 Jul 20;25(14):7937. doi: 10.3390/ijms25147937. Int J Mol Sci. 2024. PMID: 39063179 Free PMC article. Review.
-
The oral-gut microbiome axis in health and disease.Nat Rev Microbiol. 2024 Dec;22(12):791-805. doi: 10.1038/s41579-024-01075-5. Epub 2024 Jul 22. Nat Rev Microbiol. 2024. PMID: 39039286 Review.
-
Fusobacterium nucleatum elicits subspecies-specific responses in human neutrophils.Front Cell Infect Microbiol. 2024 Oct 10;14:1449539. doi: 10.3389/fcimb.2024.1449539. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 39450334 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
