

Primary osteoarthritis chondrocyte map of chromatin conformation reveals novel candidate effector genes
- PMID: 38479789
- PMCID: PMC11287644
- DOI: 10.1136/ard-2023-224945
Primary osteoarthritis chondrocyte map of chromatin conformation reveals novel candidate effector genes
Abstract
Objectives: Osteoarthritis is a complex disease with a huge public health burden. Genome-wide association studies (GWAS) have identified hundreds of osteoarthritis-associated sequence variants, but the effector genes underpinning these signals remain largely elusive. Understanding chromosome organisation in three-dimensional (3D) space is essential for identifying long-range contacts between distant genomic features (e.g., between genes and regulatory elements), in a tissue-specific manner. Here, we generate the first whole genome chromosome conformation analysis (Hi-C) map of primary osteoarthritis chondrocytes and identify novel candidate effector genes for the disease.
Methods: Primary chondrocytes collected from 8 patients with knee osteoarthritis underwent Hi-C analysis to link chromosomal structure to genomic sequence. The identified loops were then combined with osteoarthritis GWAS results and epigenomic data from primary knee osteoarthritis chondrocytes to identify variants involved in gene regulation via enhancer-promoter interactions.
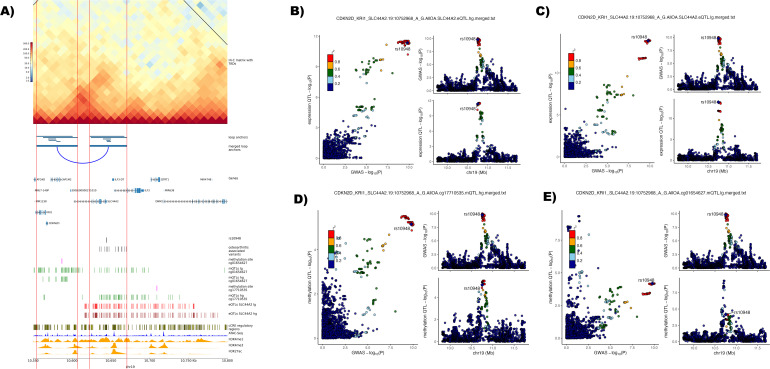
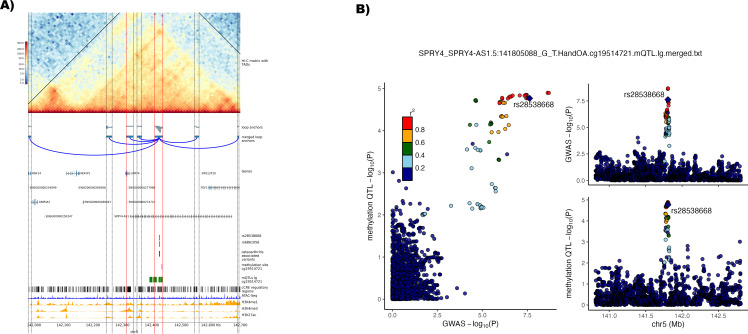
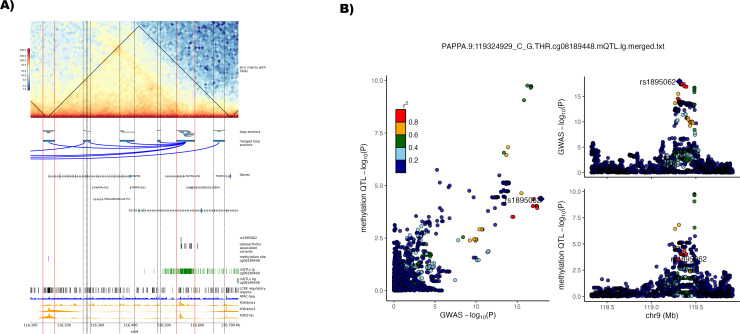
Results: We identified 345 genetic variants residing within chromatin loop anchors that are associated with 77 osteoarthritis GWAS signals. Ten of these variants reside directly in enhancer regions of 10 newly described active enhancer-promoter loops, identified with multiomics analysis of publicly available chromatin immunoprecipitation sequencing (ChIP-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) data from primary knee chondrocyte cells, pointing to two new candidate effector genes SPRY4 and PAPPA (pregnancy-associated plasma protein A) as well as further support for the gene SLC44A2 known to be involved in osteoarthritis. For example, PAPPA is directly associated with the turnover of insulin-like growth factor 1 (IGF-1) proteins, and IGF-1 is an important factor in the repair of damaged chondrocytes.
Conclusions: We have constructed the first Hi-C map of primary human chondrocytes and have made it available as a resource for the scientific community. By integrating 3D genomics with large-scale genetic association and epigenetic data, we identify novel candidate effector genes for osteoarthritis, which enhance our understanding of disease and can serve as putative high-value novel drug targets.
Keywords: chondrocytes; osteoarthritis; osteoarthritis, knee; pharmacogenetics.
© Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures


Similar articles
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Defining the optimum strategy for identifying adults and children with coeliac disease: systematic review and economic modelling.Health Technol Assess. 2022 Oct;26(44):1-310. doi: 10.3310/ZUCE8371. Health Technol Assess. 2022. PMID: 36321689 Free PMC article.
-
Discarded sequencing reads uncover natural variation in pest resistance in Thlaspi arvense.Elife. 2024 Dec 19;13:RP95510. doi: 10.7554/eLife.95510. Elife. 2024. PMID: 39699583 Free PMC article.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Exercise for osteoarthritis of the knee.Cochrane Database Syst Rev. 2015 Jan 9;1(1):CD004376. doi: 10.1002/14651858.CD004376.pub3. Cochrane Database Syst Rev. 2015. Update in: Cochrane Database Syst Rev. 2024 Dec 3;12:CD004376. doi: 10.1002/14651858.CD004376.pub4. PMID: 25569281 Free PMC article. Updated. Review.
Cited by
-
Response eQTLs, chromatin accessibility, and 3D chromatin structure in chondrocytes provide mechanistic insight into osteoarthritis risk.bioRxiv [Preprint]. 2024 Oct 31:2024.05.05.592567. doi: 10.1101/2024.05.05.592567. bioRxiv. 2024. Update in: Cell Genom. 2025 Jan 8;5(1):100738. doi: 10.1016/j.xgen.2024.100738. PMID: 38952796 Free PMC article. Updated. Preprint.
-
The metalloproteinase PAPP-A is required for IGF-dependent chondrocyte differentiation and organization.Sci Rep. 2024 Aug 29;14(1):20161. doi: 10.1038/s41598-024-71062-6. Sci Rep. 2024. PMID: 39215168 Free PMC article.
-
Bioactive Compounds and Their Chondroprotective Effects for Osteoarthritis Amelioration: A Focus on Nanotherapeutic Strategies, Epigenetic Modifications, and Gut Microbiota.Nutrients. 2024 Oct 22;16(21):3587. doi: 10.3390/nu16213587. Nutrients. 2024. PMID: 39519419 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
