

Explicit ion modeling predicts physicochemical interactions for chromatin organization
- PMID: 38289342
- PMCID: PMC10945522
- DOI: 10.7554/eLife.90073
Explicit ion modeling predicts physicochemical interactions for chromatin organization
Abstract
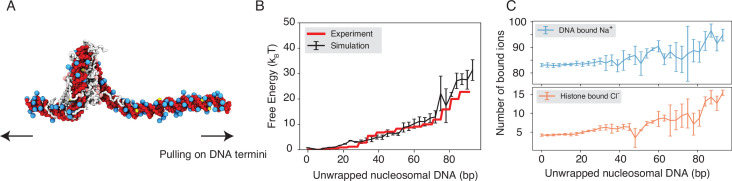
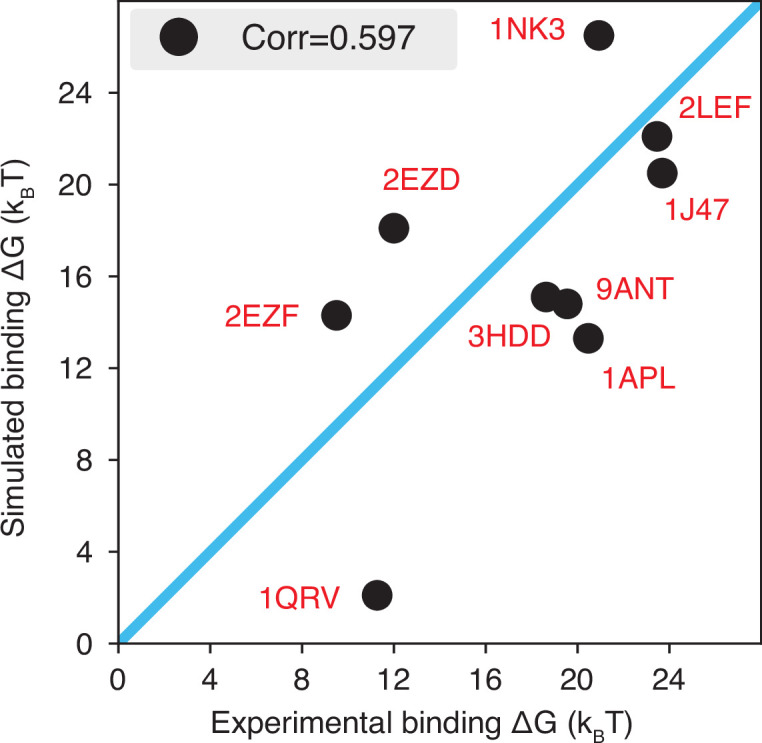
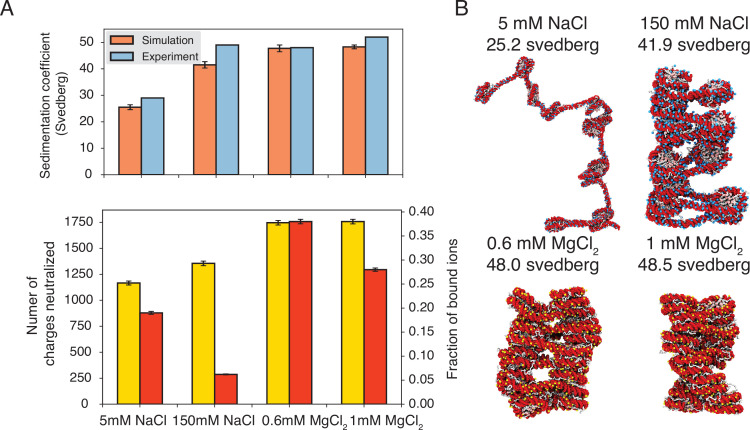
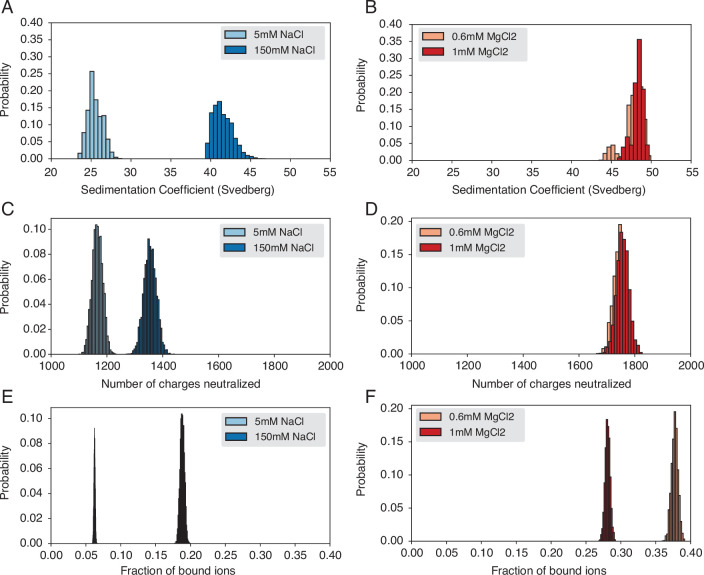
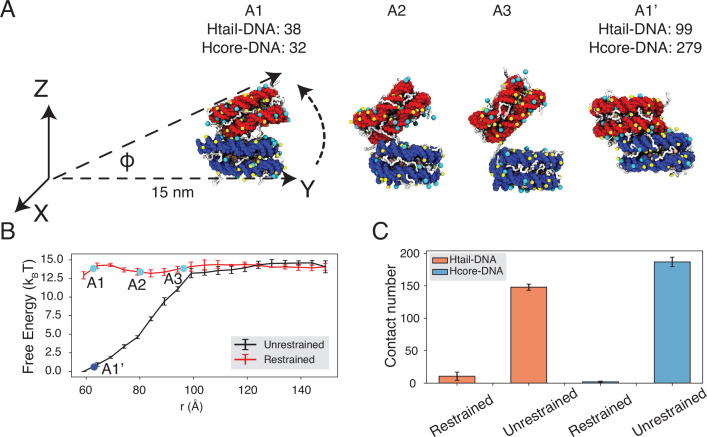
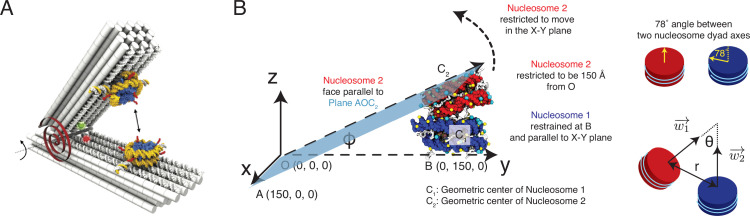
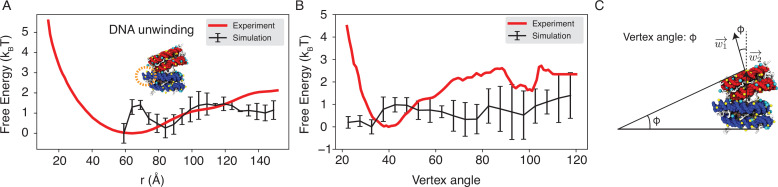
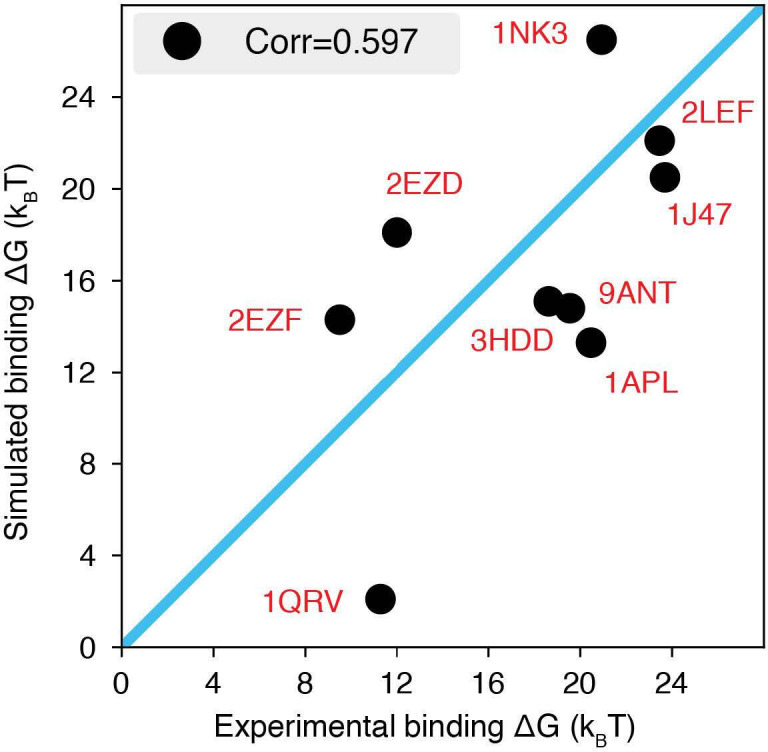
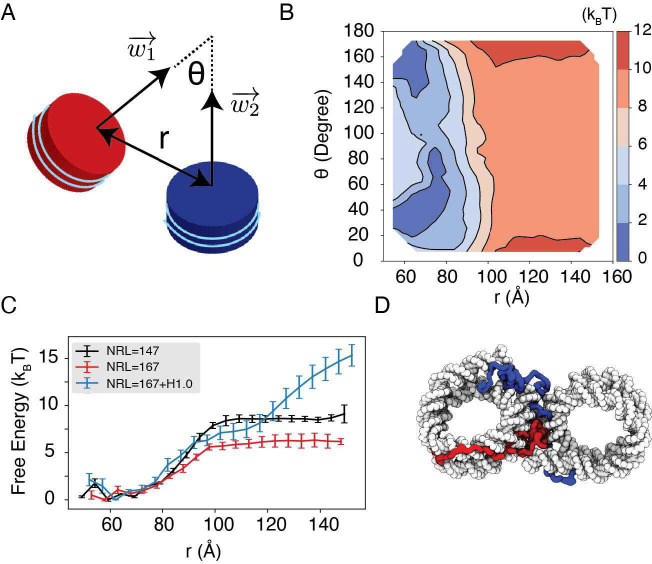
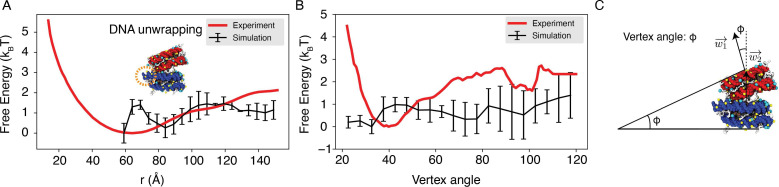
Molecular mechanisms that dictate chromatin organization in vivo are under active investigation, and the extent to which intrinsic interactions contribute to this process remains debatable. A central quantity for evaluating their contribution is the strength of nucleosome-nucleosome binding, which previous experiments have estimated to range from 2 to 14 kBT. We introduce an explicit ion model to dramatically enhance the accuracy of residue-level coarse-grained modeling approaches across a wide range of ionic concentrations. This model allows for de novo predictions of chromatin organization and remains computationally efficient, enabling large-scale conformational sampling for free energy calculations. It reproduces the energetics of protein-DNA binding and unwinding of single nucleosomal DNA, and resolves the differential impact of mono- and divalent ions on chromatin conformations. Moreover, we showed that the model can reconcile various experiments on quantifying nucleosomal interactions, providing an explanation for the large discrepancy between existing estimations. We predict the interaction strength at physiological conditions to be 9 kBT, a value that is nonetheless sensitive to DNA linker length and the presence of linker histones. Our study strongly supports the contribution of physicochemical interactions to the phase behavior of chromatin aggregates and chromatin organization inside the nucleus.
Keywords: 30 nm fiber; chromatin folding; coarse-grained modeling; explicit ions; molecular biophysics; none; structural biology.
© 2023, Lin and Zhang.
Conflict of interest statement
XL No competing interests declared, BZ Reviewing editor, eLife
Figures
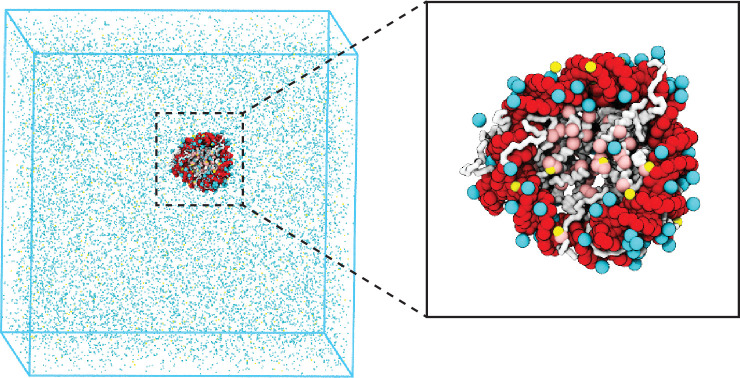
Update of
-
Explicit Ion Modeling Predicts Physicochemical Interactions for Chromatin Organization.bioRxiv [Preprint]. 2023 Nov 15:2023.05.16.541030. doi: 10.1101/2023.05.16.541030. bioRxiv. 2023. Update in: Elife. 2024 Jan 30;12:RP90073. doi: 10.7554/eLife.90073 PMID: 37293007 Free PMC article. Updated. Preprint.
Similar articles
-
Explicit Ion Modeling Predicts Physicochemical Interactions for Chromatin Organization.bioRxiv [Preprint]. 2023 Nov 15:2023.05.16.541030. doi: 10.1101/2023.05.16.541030. bioRxiv. 2023. Update in: Elife. 2024 Jan 30;12:RP90073. doi: 10.7554/eLife.90073 PMID: 37293007 Free PMC article. Updated. Preprint.
-
Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions.Proc Natl Acad Sci U S A. 2009 Aug 11;106(32):13317-22. doi: 10.1073/pnas.0903280106. Epub 2009 Jul 27. Proc Natl Acad Sci U S A. 2009. PMID: 19651606 Free PMC article.
-
A Bottom-Up Coarse-Grained Model for Nucleosome-Nucleosome Interactions with Explicit Ions.J Chem Theory Comput. 2022 Jun 14;18(6):3948-3960. doi: 10.1021/acs.jctc.2c00083. Epub 2022 May 17. J Chem Theory Comput. 2022. PMID: 35580041 Free PMC article.
-
Unraveling the multiplex folding of nucleosome chains in higher order chromatin.Essays Biochem. 2019 Apr 23;63(1):109-121. doi: 10.1042/EBC20180066. Print 2019 Apr 23. Essays Biochem. 2019. PMID: 31015386 Review.
-
Nucleosome distribution and linker DNA: connecting nuclear function to dynamic chromatin structure.Biochem Cell Biol. 2011 Feb;89(1):24-34. doi: 10.1139/O10-139. Biochem Cell Biol. 2011. PMID: 21326360 Free PMC article. Review.
Cited by
-
Transferable Implicit Solvation via Contrastive Learning of Graph Neural Networks.ACS Cent Sci. 2023 Nov 16;9(12):2286-2297. doi: 10.1021/acscentsci.3c01160. eCollection 2023 Dec 27. ACS Cent Sci. 2023. PMID: 38161379 Free PMC article.
-
Helical coiled nucleosome chromosome architectures during cell cycle progression.Proc Natl Acad Sci U S A. 2024 Oct 22;121(43):e2410584121. doi: 10.1073/pnas.2410584121. Epub 2024 Oct 14. Proc Natl Acad Sci U S A. 2024. PMID: 39401359 Free PMC article.
-
From Nucleosomes to Compartments: Physicochemical Interactions Underlying Chromatin Organization.Annu Rev Biophys. 2024 Jul;53(1):221-245. doi: 10.1146/annurev-biophys-030822-032650. Epub 2024 Jun 28. Annu Rev Biophys. 2024. PMID: 38346246 Free PMC article. Review.
-
Transferable Coarse Graining via Contrastive Learning of Graph Neural Networks.bioRxiv [Preprint]. 2023 Sep 12:2023.09.08.556923. doi: 10.1101/2023.09.08.556923. bioRxiv. 2023. Update in: ACS Cent Sci. 2023 Nov 16;9(12):2286-2297. doi: 10.1021/acscentsci.3c01160 PMID: 37745447 Free PMC article. Updated. Preprint.
-
A Molecular View into the Structure and Dynamics of Phase-Separated Chromatin.J Phys Chem B. 2024 Oct 31;128(43):10593-10603. doi: 10.1021/acs.jpcb.4c04420. Epub 2024 Oct 16. J Phys Chem B. 2024. PMID: 39413416 Free PMC article.
References
-
- Bancaud A, Wagner G, Conde E Silva N, Lavelle C, Wong H, Mozziconacci J, Barbi M, Sivolob A, Le Cam E, Mouawad L, Viovy J-L, Victor J-M, Prunell A. Nucleosome chiral transition under positive torsional stress in single chromatin fibers. Molecular Cell. 2007;27:135–147. doi: 10.1016/j.molcel.2007.05.037. - DOI - PubMed
-
- Bascom G, Schlick T. Nuclear Architecture and Dynamics. Elsevier; 2018. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
