

Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation
- PMID: 38279310
- PMCID: PMC10816320
- DOI: 10.3390/ijms25021314
Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation
Abstract
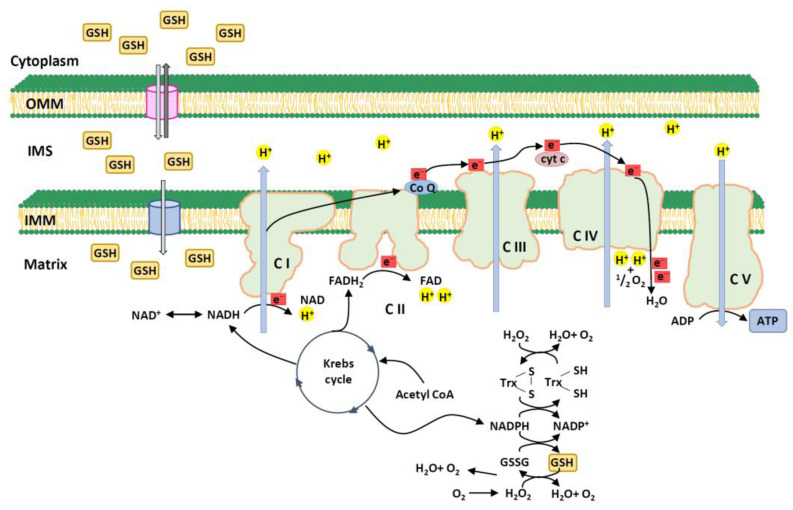
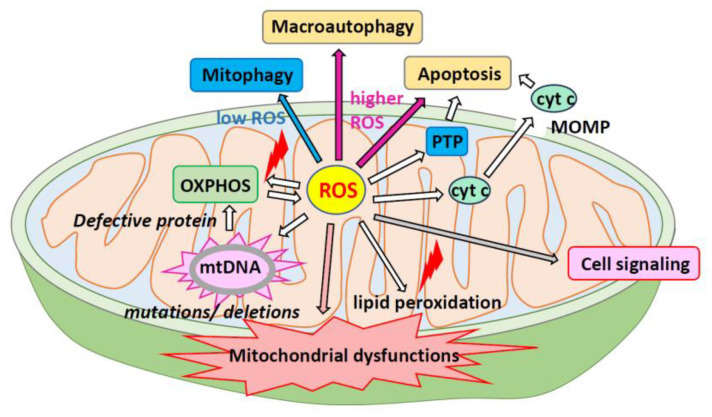
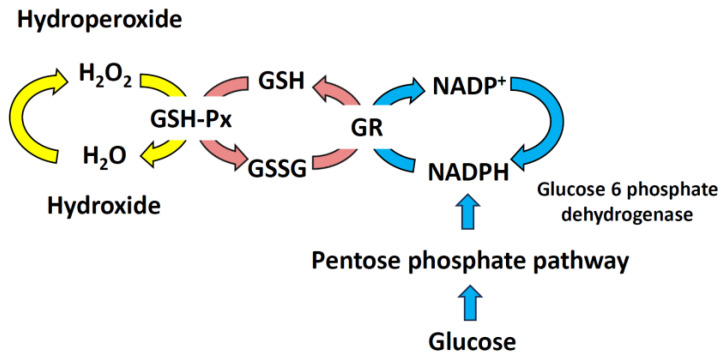
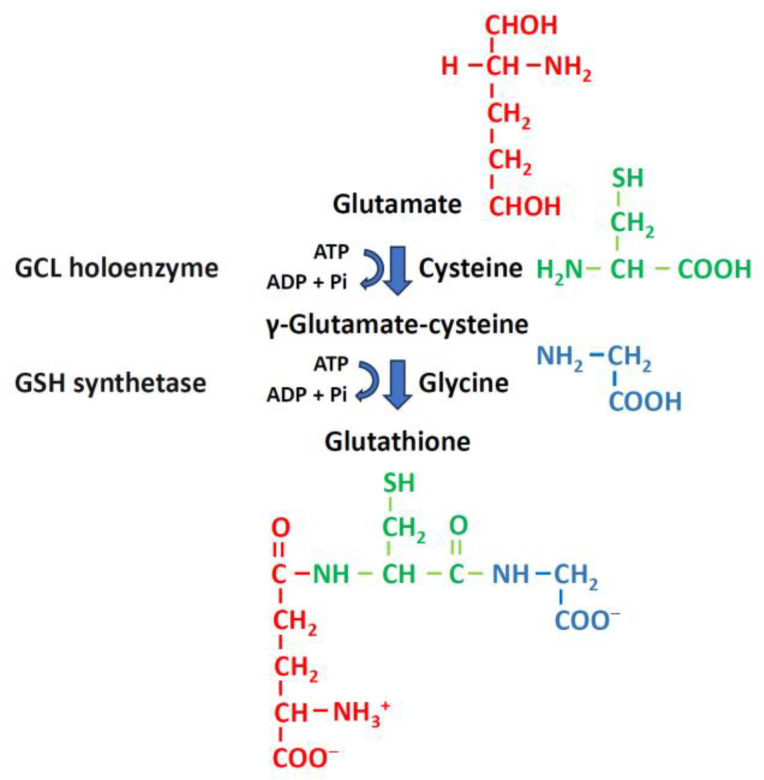
Mitochondria are critical for providing energy to maintain cell viability. Oxidative phosphorylation involves the transfer of electrons from energy substrates to oxygen to produce adenosine triphosphate. Mitochondria also regulate cell proliferation, metastasis, and deterioration. The flow of electrons in the mitochondrial respiratory chain generates reactive oxygen species (ROS), which are harmful to cells at high levels. Oxidative stress caused by ROS accumulation has been associated with an increased risk of cancer, and cardiovascular and liver diseases. Glutathione (GSH) is an abundant cellular antioxidant that is primarily synthesized in the cytoplasm and delivered to the mitochondria. Mitochondrial glutathione (mGSH) metabolizes hydrogen peroxide within the mitochondria. A long-term imbalance in the ratio of mitochondrial ROS to mGSH can cause cell dysfunction, apoptosis, necroptosis, and ferroptosis, which may lead to disease. This study aimed to review the physiological functions, anabolism, variations in organ tissue accumulation, and delivery of GSH to the mitochondria and the relationships between mGSH levels, the GSH/GSH disulfide (GSSG) ratio, programmed cell death, and ferroptosis. We also discuss diseases caused by mGSH deficiency and related therapeutics.
Keywords: GSH deficiency; GSH/GSSG; glutathione; mitochondria; oxidative phosphorylation; programmed cell death; reactive oxygen species (ROS).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases.Biochim Biophys Acta. 2015 Jun-Jul;1847(6-7):514-25. doi: 10.1016/j.bbabio.2015.02.012. Epub 2015 Feb 19. Biochim Biophys Acta. 2015. PMID: 25701705 Free PMC article.
-
Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria.Redox Biol. 2016 Aug;8:110-8. doi: 10.1016/j.redox.2015.12.010. Epub 2015 Dec 31. Redox Biol. 2016. PMID: 26773874 Free PMC article. Review.
-
Mitochondrial glutathione: features, regulation and role in disease.Biochim Biophys Acta. 2013 May;1830(5):3317-28. doi: 10.1016/j.bbagen.2012.10.018. Epub 2012 Oct 30. Biochim Biophys Acta. 2013. PMID: 23123815 Free PMC article. Review.
-
Mitochondrial glutathione and oxidative stress: implications for pulmonary oxygen toxicity in premature infants.Mol Genet Metab. 2000 Sep-Oct;71(1-2):352-8. doi: 10.1006/mgme.2000.3063. Mol Genet Metab. 2000. PMID: 11001827 Review.
-
Computational insights into the role of glutathione in oxidative stress.Curr Neurovasc Res. 2013 May;10(2):185-94. doi: 10.2174/1567202611310020011. Curr Neurovasc Res. 2013. PMID: 23469953 Review.
Cited by
-
Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases.Antioxidants (Basel). 2024 Sep 25;13(10):1163. doi: 10.3390/antiox13101163. Antioxidants (Basel). 2024. PMID: 39456418 Free PMC article. Review.
-
Research progress on ferroptosis in the pathogenesis and treatment of neurodegenerative diseases.Front Cell Neurosci. 2024 Mar 7;18:1359453. doi: 10.3389/fncel.2024.1359453. eCollection 2024. Front Cell Neurosci. 2024. PMID: 38515787 Free PMC article. Review.
-
Building a Human Ovarian Antioxidant ceRNA Network "OvAnOx": A Bioinformatic Perspective for Research on Redox-Related Ovarian Functions and Dysfunctions.Antioxidants (Basel). 2024 Sep 12;13(9):1101. doi: 10.3390/antiox13091101. Antioxidants (Basel). 2024. PMID: 39334761 Free PMC article. Review.
-
Glutathione in HIV-Associated Neurocognitive Disorders.Curr Issues Mol Biol. 2024 May 31;46(6):5530-5549. doi: 10.3390/cimb46060330. Curr Issues Mol Biol. 2024. PMID: 38921002 Free PMC article. Review.
-
Nebulized Glutathione as a Key Antioxidant for the Treatment of Oxidative Stress in Neurodegenerative Conditions.Nutrients. 2024 Jul 31;16(15):2476. doi: 10.3390/nu16152476. Nutrients. 2024. PMID: 39125356 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
