

Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction
- PMID: 38257371
- PMCID: PMC10818339
- DOI: 10.3390/molecules29020453
Identification of GM1-Ganglioside Secondary Accumulation in Fibroblasts from Neuropathic Gaucher Patients and Effect of a Trivalent Trihydroxypiperidine Iminosugar Compound on Its Storage Reduction
Abstract
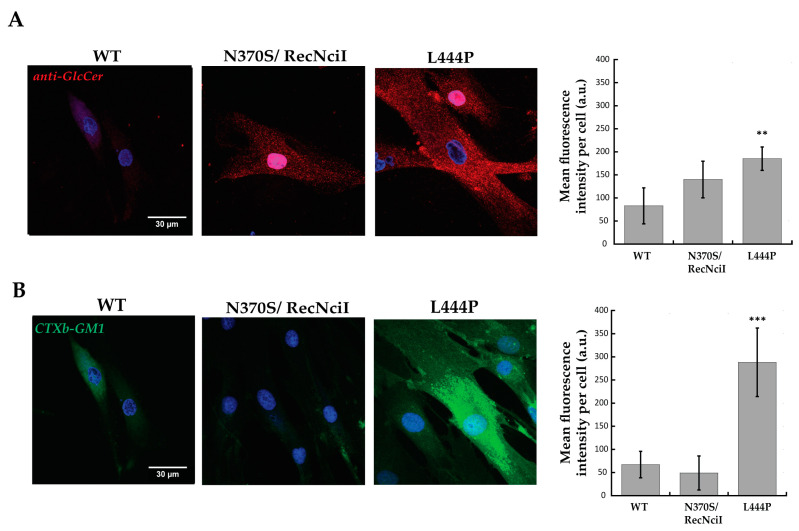
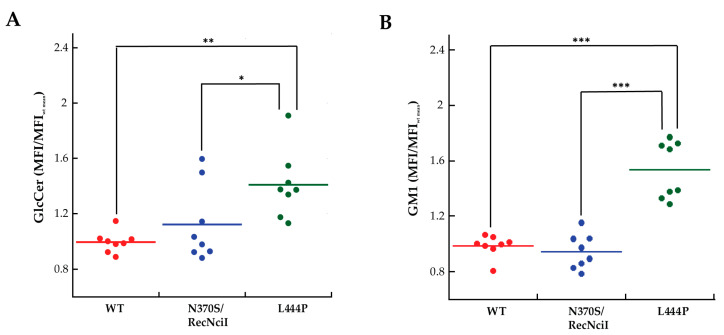
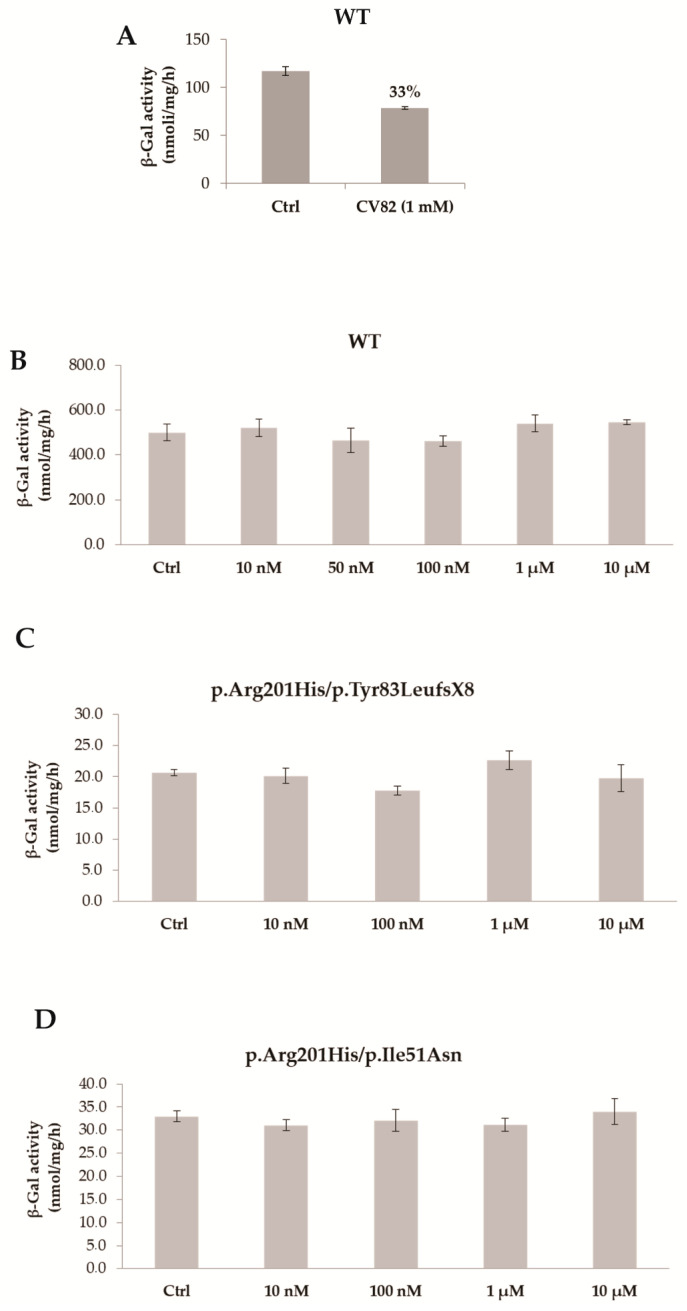
Gaucher disease (GD) is a rare genetic metabolic disorder characterized by a dysfunction of the lysosomal glycoside hydrolase glucocerebrosidase (GCase) due to mutations in the gene GBA1, leading to the cellular accumulation of glucosylceramide (GlcCer). While most of the current research focuses on the primary accumulated material, lesser attention has been paid to secondary storage materials and their reciprocal intertwining. By using a novel approach based on flow cytometry and fluorescent labelling, we monitored changes in storage materials directly in fibroblasts derived from GD patients carrying N370S/RecNcil and homozygous L444P or R131C mutations with respect to wild type. In L444P and R131C fibroblasts, we detected not only the primary accumulation of GlcCer accumulation but also a considerable secondary increase in GM1 storage, comparable with the one observed in infantile patients affected by GM1 gangliosidosis. In addition, the ability of a trivalent trihydroxypiperidine iminosugar compound (CV82), which previously showed good pharmacological chaperone activity on GCase enzyme, to reduce the levels of storage materials in L444P and R131C fibroblasts was tested. Interestingly, treatment with different concentrations of CV82 led to a significant reduction in GM1 accumulation only in L444P fibroblasts, without significantly affecting GlcCer levels. The compound CV82 was selective against the GCase enzyme with respect to the β-Galactosidase enzyme, which was responsible for the catabolism of GM1 ganglioside. The reduction in GM1-ganglioside level cannot be therefore ascribed to a direct action of CV82 on β-Galactosidase enzyme, suggesting that GM1 decrease is rather related to other unknown mechanisms that follow the direct action of CV82 on GCase. In conclusion, this work indicates that the tracking of secondary storages can represent a key step for a better understanding of the pathways involved in the severity of GD, also underlying the importance of developing drugs able to reduce both primary and secondary storage-material accumulations in GD.
Keywords: GM1; flow cytometry; glucocerebrosidase; glucosylceramide; lysosome; metabolic disorders; pharmacological chaperones.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


Similar articles
-
Novel beta-glucocerebrosidase chaperone compounds identified from cell-based screening reduce pathologically accumulated glucosylsphingosine in iPS-derived neuronal cells.SLAS Discov. 2023 Oct;28(7):344-349. doi: 10.1016/j.slasd.2023.06.002. Epub 2023 Jun 25. SLAS Discov. 2023. PMID: 37369311
-
Glucosylceramide mimics: highly potent GCase inhibitors and selective pharmacological chaperones for mutations associated with types 1 and 2 Gaucher disease.ChemMedChem. 2013 Nov;8(11):1805-17. doi: 10.1002/cmdc.201300327. Epub 2013 Oct 2. ChemMedChem. 2013. PMID: 24115322
-
3,4,5-Trihydroxypiperidine Based Multivalent Glucocerebrosidase (GCase) Enhancers.Chembiochem. 2022 Jun 3;23(11):e202200077. doi: 10.1002/cbic.202200077. Epub 2022 Apr 7. Chembiochem. 2022. PMID: 35322924 Free PMC article.
-
Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease.Brain Dev. 2013 Jun;35(6):515-23. doi: 10.1016/j.braindev.2012.12.002. Epub 2013 Jan 3. Brain Dev. 2013. PMID: 23290321 Review.
-
Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease.Adv Drug Deliv Rev. 2022 Aug;187:114402. doi: 10.1016/j.addr.2022.114402. Epub 2022 Jun 25. Adv Drug Deliv Rev. 2022. PMID: 35764179 Review.
References
-
- Zimran A., Elstein D. Management of Gaucher Disease: Enzyme Replacement Therapy. Pediatr. Endocrinol. Rev. 2014;12((Suppl. S1)):82–87. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
