

The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome
- PMID: 38254639
- PMCID: PMC10813406
- DOI: 10.3390/biom14010039
The Role of the Complement System in the Pathogenesis of Infectious Forms of Hemolytic Uremic Syndrome
Abstract
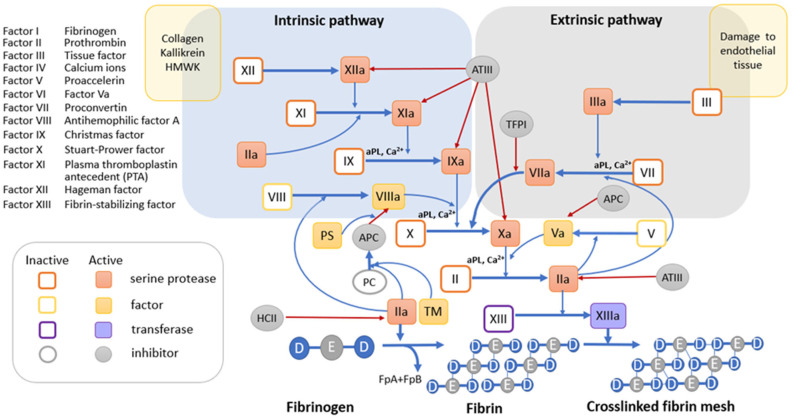
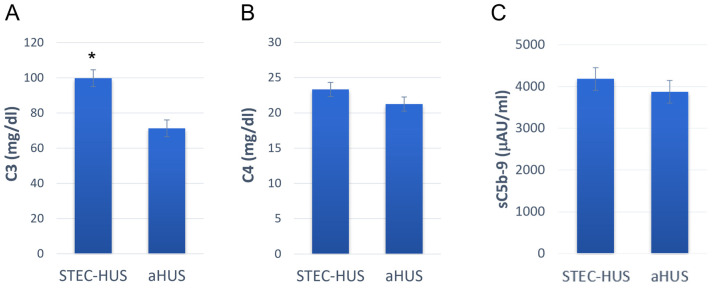
Hemolytic uremic syndrome (HUS) is an acute disease and the most common cause of childhood acute renal failure. HUS is characterized by a triad of symptoms: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. In most of the cases, HUS occurs as a result of infection caused by Shiga toxin-producing microbes: hemorrhagic Escherichia coli and Shigella dysenteriae type 1. They account for up to 90% of all cases of HUS. The remaining 10% of cases grouped under the general term atypical HUS represent a heterogeneous group of diseases with similar clinical signs. Emerging evidence suggests that in addition to E. coli and S. dysenteriae type 1, a variety of bacterial and viral infections can cause the development of HUS. In particular, infectious diseases act as the main cause of aHUS recurrence. The pathogenesis of most cases of atypical HUS is based on congenital or acquired defects of complement system. This review presents summarized data from recent studies, suggesting that complement dysregulation is a key pathogenetic factor in various types of infection-induced HUS. Separate links in the complement system are considered, the damage of which during bacterial and viral infections can lead to complement hyperactivation following by microvascular endothelial injury and development of acute renal failure.
Keywords: Escherichia coli; STEC-HUS; Shiga toxin; acute renal failure; complement system; eculizumab; endothelium; hemolytic anemia; hemolytic uremic syndrome; pathogenesis; thrombocytopenia; thrombotic microangiopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Hemolytic uremic syndrome: differential diagnosis with the onset of inflammatory bowel diseases.Acta Biomed. 2018 Dec 17;89(9-S):153-157. doi: 10.23750/abm.v89i9-S.7911. Acta Biomed. 2018. PMID: 30561409 Free PMC article.
-
Hemolytic uremic syndrome in children.Minerva Pediatr. 2016 Dec;68(6):441-455. Minerva Pediatr. 2016. PMID: 27768015 Review.
-
Hemolytic uremic syndrome.Semin Immunopathol. 2014 Jul;36(4):399-420. doi: 10.1007/s00281-014-0416-x. Epub 2014 Feb 14. Semin Immunopathol. 2014. PMID: 24526222 Review.
-
Where are we with haemolytic uremic syndrome?Med Clin (Barc). 2018 Oct 23;151(8):329-335. doi: 10.1016/j.medcli.2018.02.016. Epub 2018 Apr 24. Med Clin (Barc). 2018. PMID: 29699703 Review. English, Spanish.
-
Thrombotic microangiopathies: thrombotic thrombocytopenic purpura / hemolytic uremic syndrome.J Bras Nefrol. 2010 Jul-Sep;32(3):303-15. J Bras Nefrol. 2010. PMID: 21103695
Cited by
-
Immunity and Coagulation in COVID-19.Int J Mol Sci. 2024 Oct 19;25(20):11267. doi: 10.3390/ijms252011267. Int J Mol Sci. 2024. PMID: 39457048 Free PMC article. Review.
-
Exploring the Intersection of Atypical Hemolytic Uremic Syndrome and Substance Use: A Comprehensive Narrative Review.Cureus. 2024 Oct 7;16(10):e71019. doi: 10.7759/cureus.71019. eCollection 2024 Oct. Cureus. 2024. PMID: 39507167 Free PMC article. Review.
References
-
- Fakhouri F., Fila M., Hummel A., Ribes D., Sellier-Leclerc A.L., Ville S., Pouteil-Noble C., Coindre J.P., Le Quintrec M., Rondeau E., et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood. 2021;137:2438–2449. doi: 10.1182/blood.2020009280. - DOI - PubMed
-
- Loirat C., Fakhouri F., Ariceta G., Besbas N., Bitzan M., Bjerre A., Coppo R., Emma F., Johnson S., Karpman D., et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016;31:15–39. doi: 10.1007/s00467-015-3076-8. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
