

Tonic ErbB signaling underlies TGFβ-induced activation of ERK and is required for lens cell epithelial to myofibroblast transition
- PMID: 38170570
- PMCID: PMC10916858
- DOI: 10.1091/mbc.E23-07-0294
Tonic ErbB signaling underlies TGFβ-induced activation of ERK and is required for lens cell epithelial to myofibroblast transition
Abstract
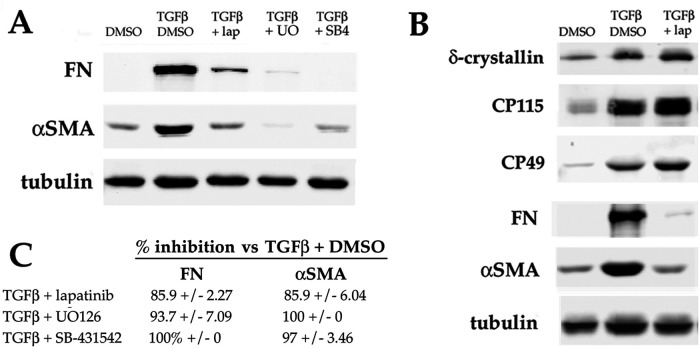
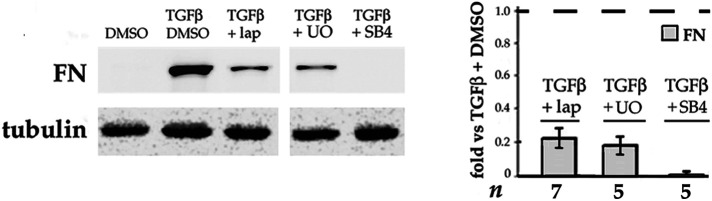
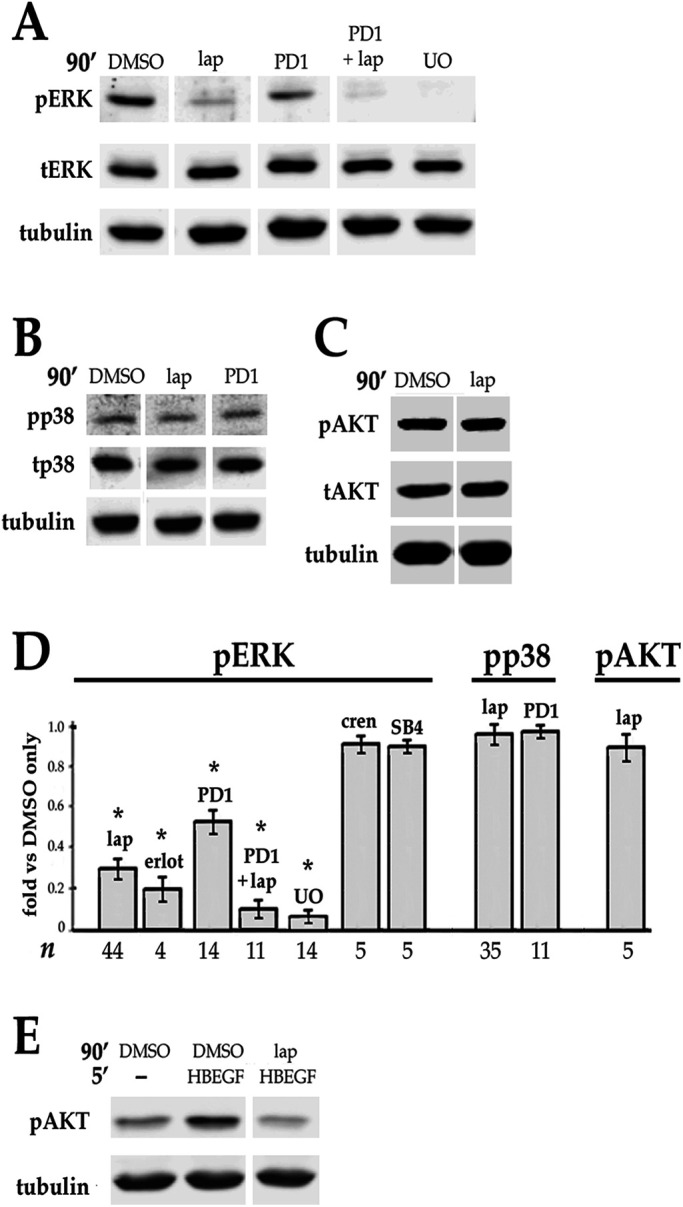
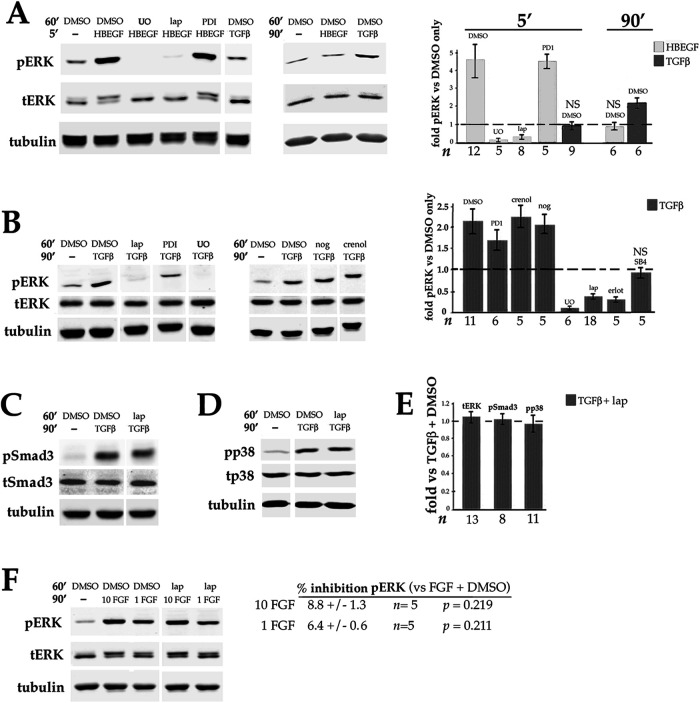
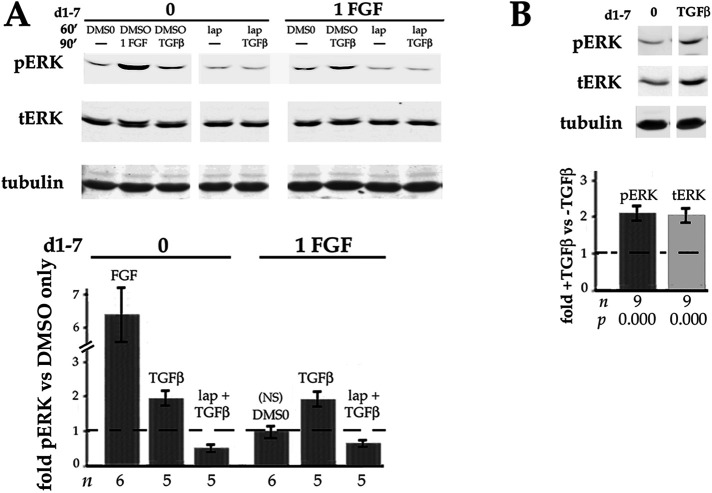
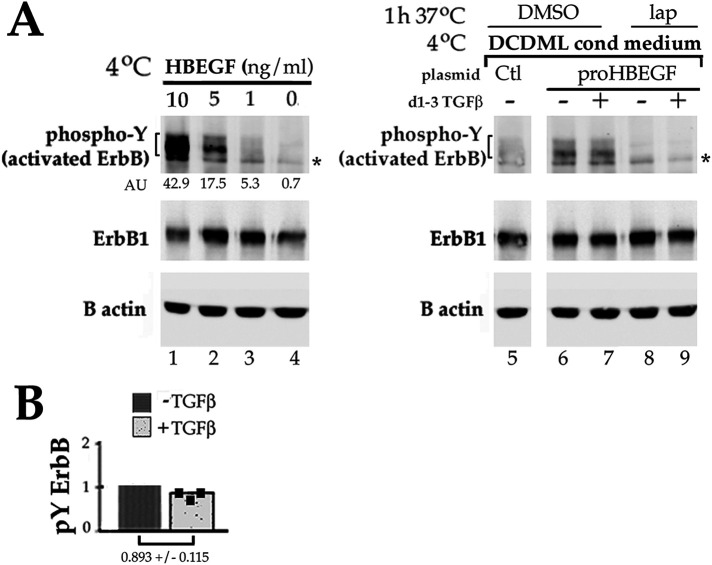
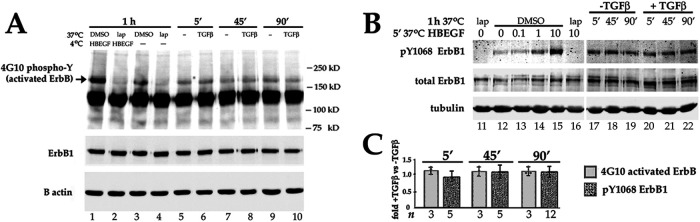
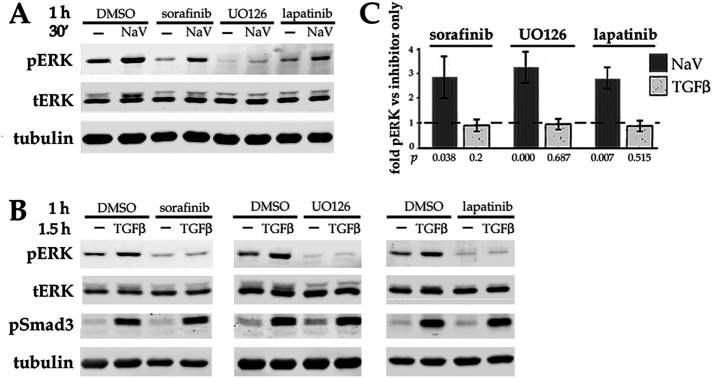
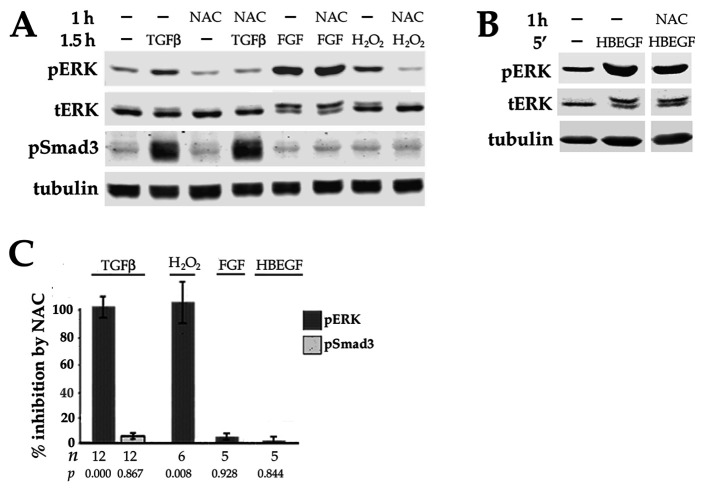
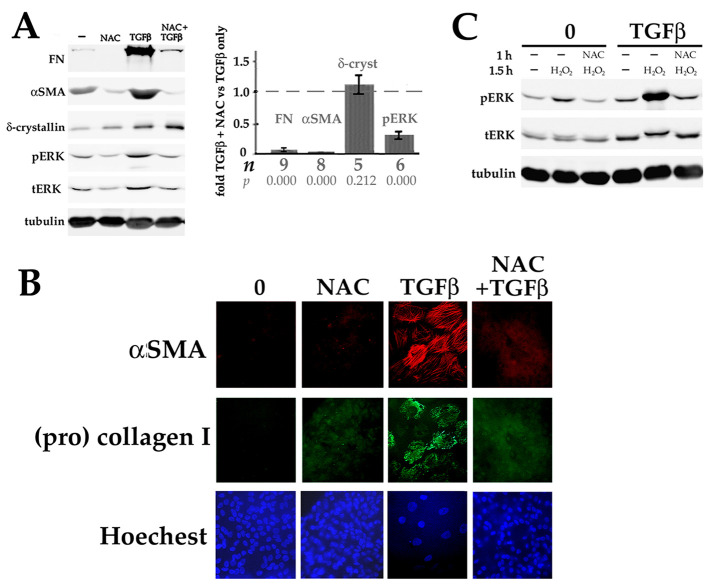
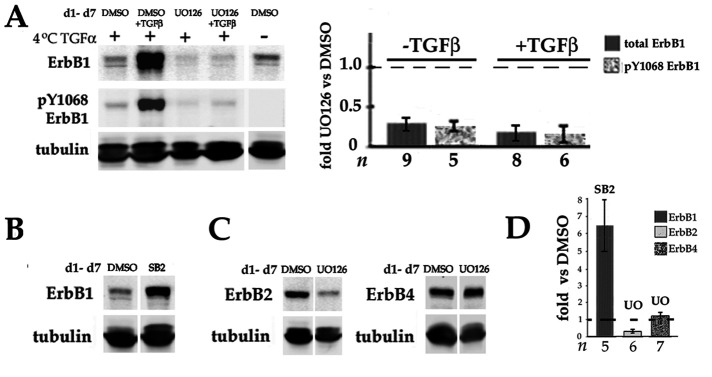
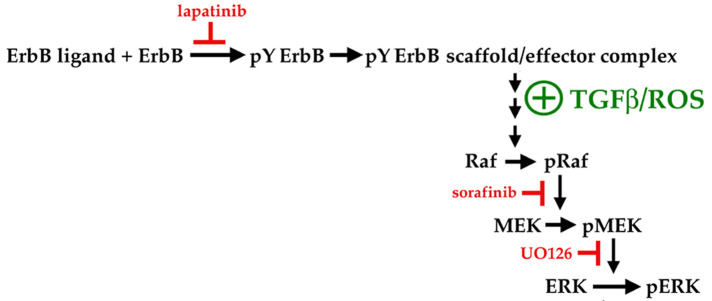
Fibrosis is a major, but incompletely understood, component of many diseases. The most common vision-disrupting complication of cataract surgery involves differentiation of residual lens cells into myofibroblasts. In serum-free primary cultures of lens epithelial cells (DCDMLs), inhibitors of either ERK or of ErbB signaling prevent TGFβ from upregulating both early (fibronectin) and late (αSMA) markers of myofibroblast differentiation. TGFβ stimulates ERK in DCDMLs within 1.5 h. Kinase inhibitors of ErbBs, but not of several other growth factor receptors in lens cells, reduce phospho ERK to below basal levels in the absence or presence of TGFβ. This effect is attributable to constitutive ErbB activity playing a major role in regulating the basal levels pERK. Additional studies support a model in which TGFβ-generated reactive oxygen species serve to indirectly amplify ERK signaling downstream of tonically active ErbBs to mediate myofibroblast differentiation. ERK activity is in turn essential for expression of ErbB1 and ErbB2, major inducers of ERK signaling. By mechanistically linking TGFβ, ErbB, and ERK signaling to myofibroblast differentiation, our data elucidate a new role for ErbBs in fibrosis and reveal a novel mode by which TGFβ directs lens cell fate.
Figures
Similar articles
-
TGFβ overcomes FGF-induced transinhibition of EGFR in lens cells to enable fibrotic secondary cataract.Mol Biol Cell. 2024 Jun 1;35(6):ar75. doi: 10.1091/mbc.E24-01-0040. Epub 2024 Apr 10. Mol Biol Cell. 2024. PMID: 38598298 Free PMC article.
-
ErbBs in Lens Cell Fibrosis and Secondary Cataract.Invest Ophthalmol Vis Sci. 2023 Jul 3;64(10):6. doi: 10.1167/iovs.64.10.6. Invest Ophthalmol Vis Sci. 2023. PMID: 37418274 Free PMC article.
-
Dual function of TGFβ in lens epithelial cell fate: implications for secondary cataract.Mol Biol Cell. 2017 Apr 1;28(7):907-921. doi: 10.1091/mbc.E16-12-0865. Epub 2017 Feb 16. Mol Biol Cell. 2017. PMID: 28209733 Free PMC article.
-
Fibrosis in the lens. Sprouty regulation of TGFβ-signaling prevents lens EMT leading to cataract.Exp Eye Res. 2016 Jan;142:92-101. doi: 10.1016/j.exer.2015.02.004. Epub 2015 May 21. Exp Eye Res. 2016. PMID: 26003864 Free PMC article. Review.
-
Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation.Cells Tissues Organs. 2005;179(1-2):43-55. doi: 10.1159/000084508. Cells Tissues Organs. 2005. PMID: 15942192 Review.
Cited by
-
TGFβ overcomes FGF-induced transinhibition of EGFR in lens cells to enable fibrotic secondary cataract.Mol Biol Cell. 2024 Jun 1;35(6):ar75. doi: 10.1091/mbc.E24-01-0040. Epub 2024 Apr 10. Mol Biol Cell. 2024. PMID: 38598298 Free PMC article.
References
-
- Apple DJ, Ram J, Foster A, Peng Q (2000). Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol 45(Suppl 1), S1–S196. - PubMed
-
- Awasthi N, Guo S, Wagner BJ (2009). Posterior capsular opacification: a problem reduced but not yet eradicated. Arch Ophthalmol 127, 555–562. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
