

N-acetyltransferase 10 regulates alphavirus replication via N4-acetylcytidine (ac4C) modification of the lymphocyte antigen six family member E (LY6E) mRNA
- PMID: 38169284
- PMCID: PMC10805074
- DOI: 10.1128/jvi.01350-23
N-acetyltransferase 10 regulates alphavirus replication via N4-acetylcytidine (ac4C) modification of the lymphocyte antigen six family member E (LY6E) mRNA
Abstract
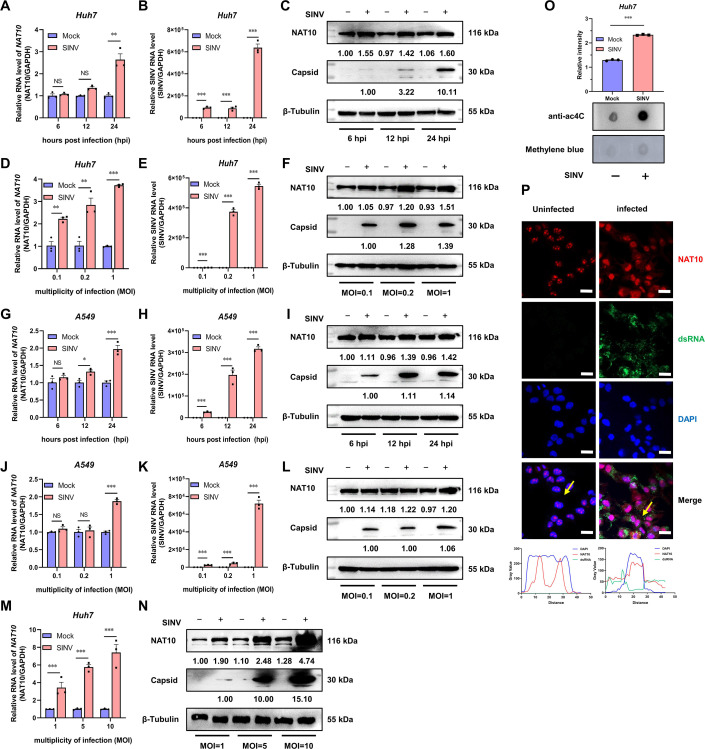
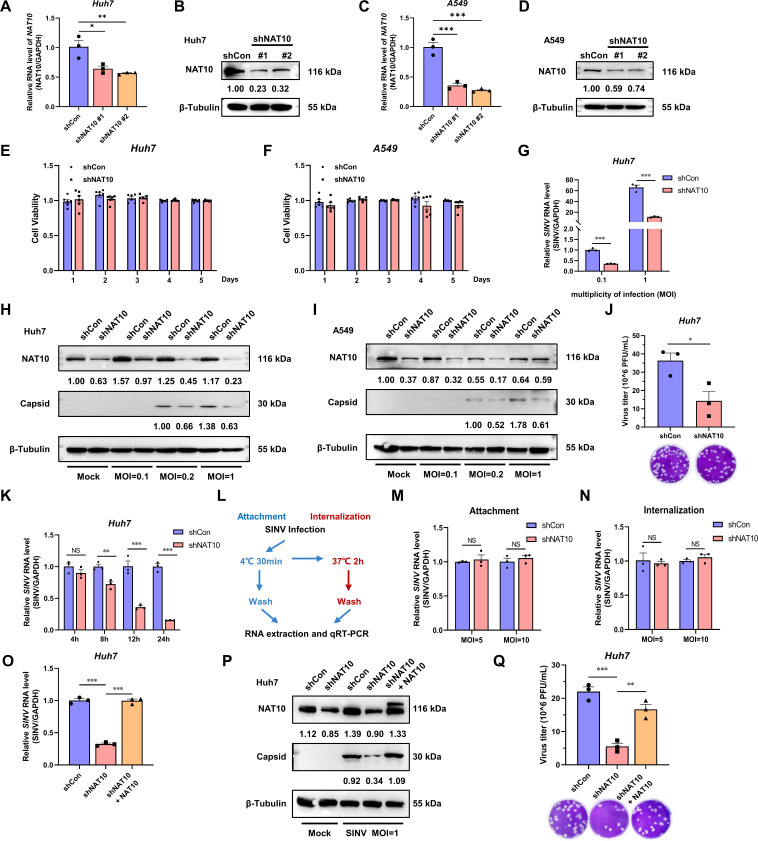
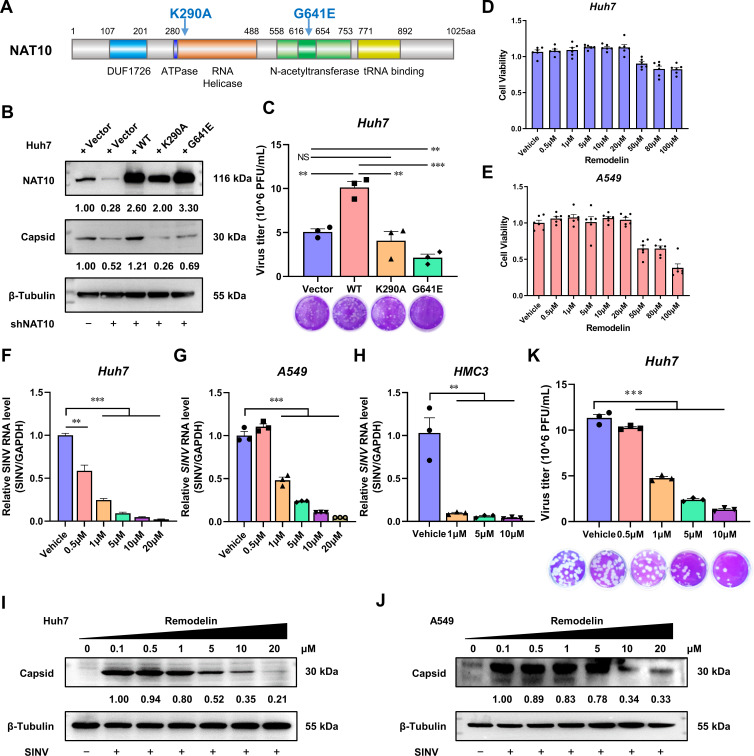
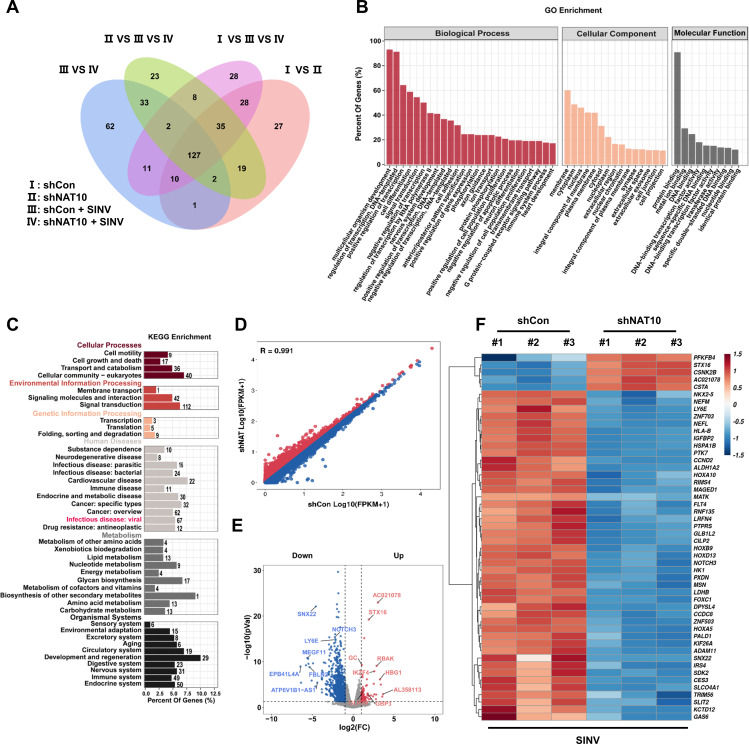
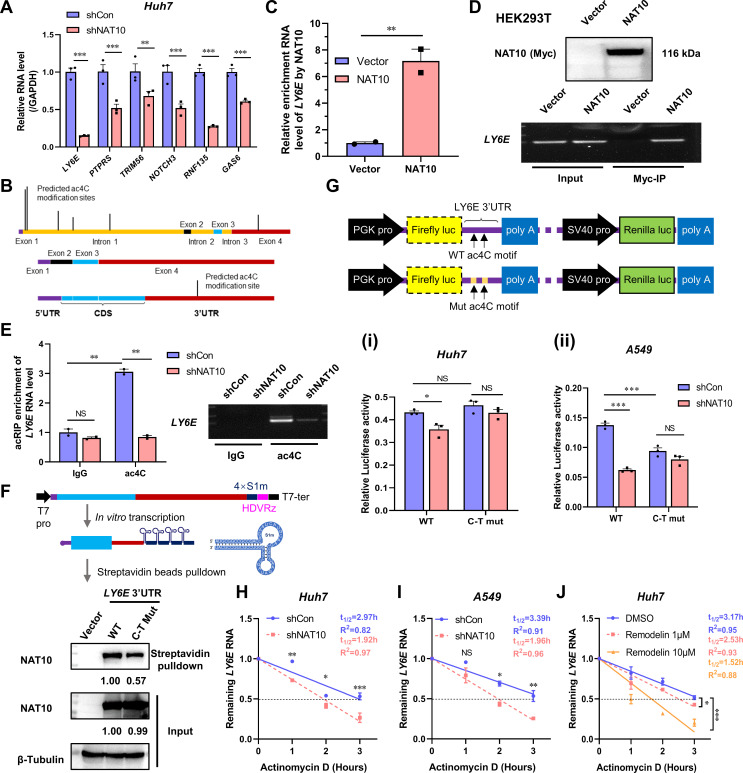
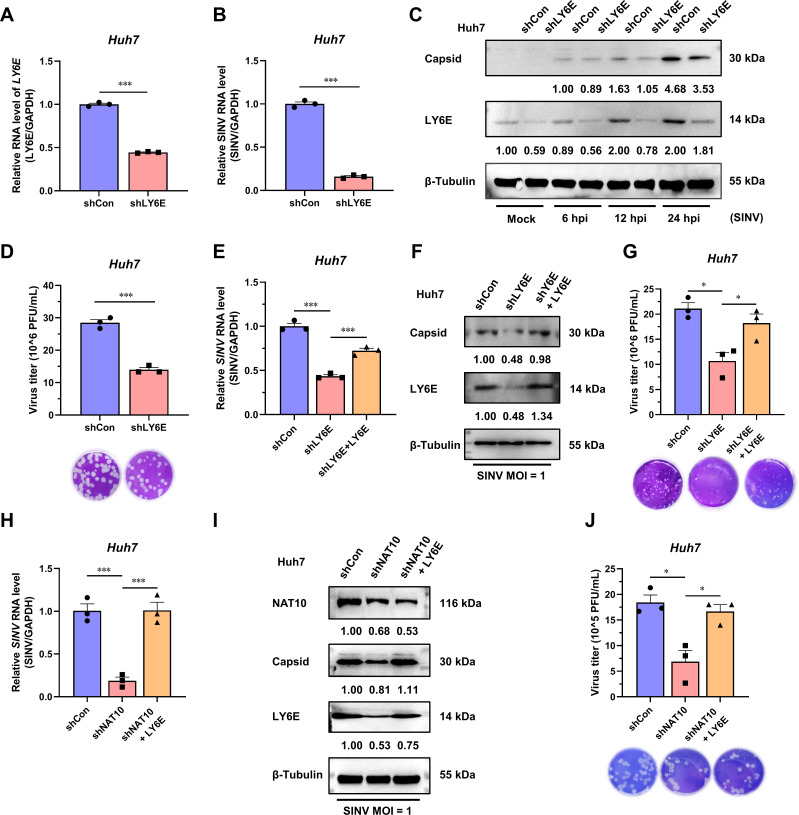
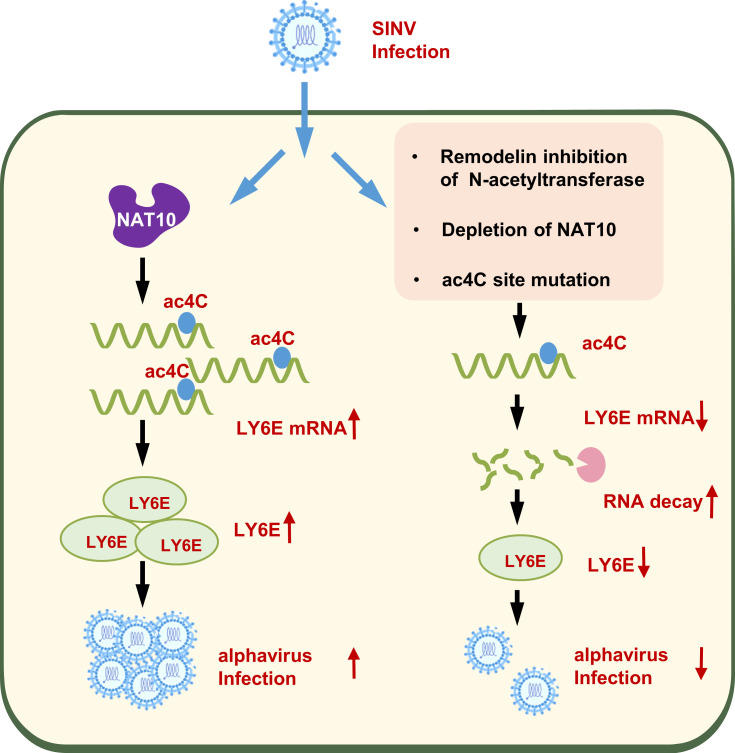
Epitranscriptomic RNA modifications can regulate the stability of mRNA and affect cellular and viral RNA functions. The N4-acetylcytidine (ac4C) modification in the RNA viral genome was recently found to promote viral replication; however, the mechanism by which RNA acetylation in the host mRNA regulates viral replication remains unclear. To help elucidate this mechanism, the roles of N-acetyltransferase 10 (NAT10) and ac4C during the infection and replication processes of the alphavirus, Sindbis virus (SINV), were investigated. Cellular NAT10 was upregulated, and ac4C modifications were promoted after alphavirus infection, while the loss of NAT10 or inhibition of its N-acetyltransferase activity reduced alphavirus replication. The NAT10 enhanced alphavirus replication as it helped to maintain the stability of lymphocyte antigen six family member E mRNA, which is a multifunctional interferon-stimulated gene that promotes alphavirus replication. The ac4C modification was thus found to have a non-conventional role in the virus life cycle through regulating host mRNA stability instead of viral mRNA, and its inhibition could be a potential target in the development of new alphavirus antivirals.IMPORTANCEThe role of N4-acetylcytidine (ac4C) modification in host mRNA and virus replication is not yet fully understood. In this study, the role of ac4C in the regulation of Sindbis virus (SINV), a prototype alphavirus infection, was investigated. SINV infection results in increased levels of N-acetyltransferase 10 (NAT10) and increases the ac4C modification level of cellular RNA. The NAT10 was found to positively regulate SINV infection in an N-acetyltransferase activity-dependent manner. Mechanistically, the NAT10 modifies lymphocyte antigen six family member E (LY6E) mRNA-the ac4C modification site within the 3'-untranslated region (UTR) of LY6E mRNA, which is essential for its translation and stability. The findings of this study demonstrate that NAT10 regulated mRNA stability and translation efficiency not only through the 5'-UTR or coding sequence but also via the 3'-UTR region. The ac4C modification of host mRNA stability instead of viral mRNA impacting the viral life cycle was thus identified, indicating that the inhibition of ac4C could be a potential target when developing alphavirus antivirals.
Keywords: N-acetyltransferase 10 (NAT10); N4-acetylcytidine (ac4C); Sindbis virus (SINV); alphavirus; lymphocyte antigen 6 family member E (LY6E).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Targeting N4-acetylcytidine suppresses hepatocellular carcinoma progression by repressing eEF2-mediated HMGB2 mRNA translation.Cancer Commun (Lond). 2024 Sep;44(9):1018-1041. doi: 10.1002/cac2.12595. Epub 2024 Jul 19. Cancer Commun (Lond). 2024. PMID: 39030964 Free PMC article.
-
NAT10 promotes liver lipogenesis in mouse through N4-acetylcytidine modification of Srebf1 and Scap mRNA.Lipids Health Dis. 2024 Nov 11;23(1):368. doi: 10.1186/s12944-024-02360-1. Lipids Health Dis. 2024. PMID: 39529018 Free PMC article.
-
NAT10 promotes vascular remodelling via mRNA ac4C acetylation.Eur Heart J. 2025 Jan 16;46(3):288-304. doi: 10.1093/eurheartj/ehae707. Eur Heart J. 2025. PMID: 39453784
-
Mechanisms of NAT10 as ac4C writer in diseases.Mol Ther Nucleic Acids. 2023 Apr 3;32:359-368. doi: 10.1016/j.omtn.2023.03.023. eCollection 2023 Jun 13. Mol Ther Nucleic Acids. 2023. PMID: 37128278 Free PMC article. Review.
-
NAT10 and cytidine acetylation in mRNA: intersecting paths in development and disease.Curr Opin Genet Dev. 2024 Aug;87:102207. doi: 10.1016/j.gde.2024.102207. Epub 2024 May 30. Curr Opin Genet Dev. 2024. PMID: 38820741 Review.
Cited by
-
Differing Transcriptomic Responses in High Titer versus Low Titer Aedes aegypti Mosquitoes after Oral Infection with Sindbis Virus.Viruses. 2024 Sep 19;16(9):1487. doi: 10.3390/v16091487. Viruses. 2024. PMID: 39339963 Free PMC article.
References
-
- Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD, Fox SD, Zengeya TT, Andresson T, Meier JL, Coller J, Oberdoerffer S. 2018. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175:1872–1886. doi:10.1016/j.cell.2018.10.030 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
- 81871649/MOST | National Natural Science Foundation of China (NSFC)
- 82172272/MOST | National Natural Science Foundation of China (NSFC)
- 2021JSTS10/Airforce Medical University supporting grant
- 2016YFC1202903/MOST | National Key Research and Development Program of China (NKPs)
- 2022YFC2604200/MOST | National Key Research and Development Program of China (NKPs)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
