

This is a preprint.
Population-scale skeletal muscle single-nucleus multi-omic profiling reveals extensive context specific genetic regulation
- PMID: 38168419
- PMCID: PMC10760134
- DOI: 10.1101/2023.12.15.571696
Population-scale skeletal muscle single-nucleus multi-omic profiling reveals extensive context specific genetic regulation
Abstract
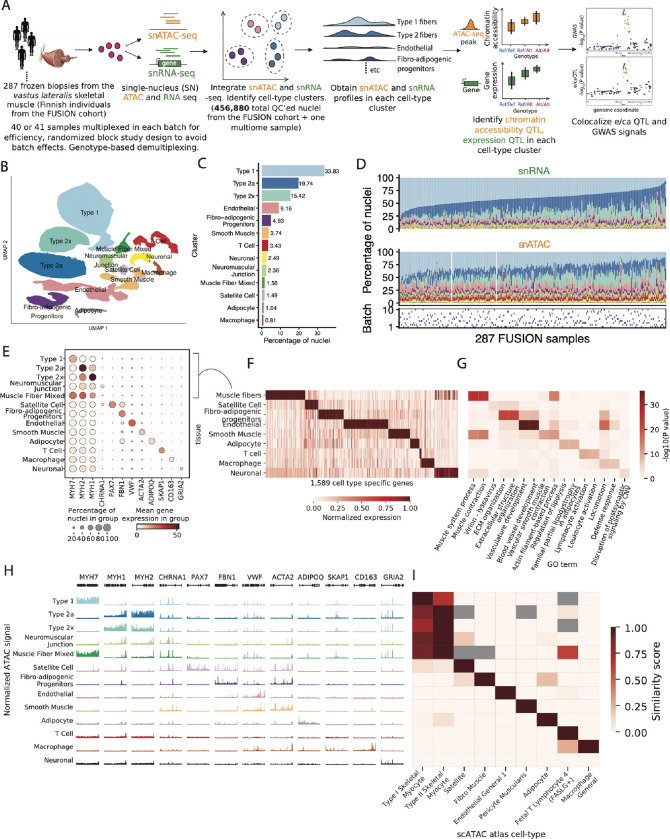
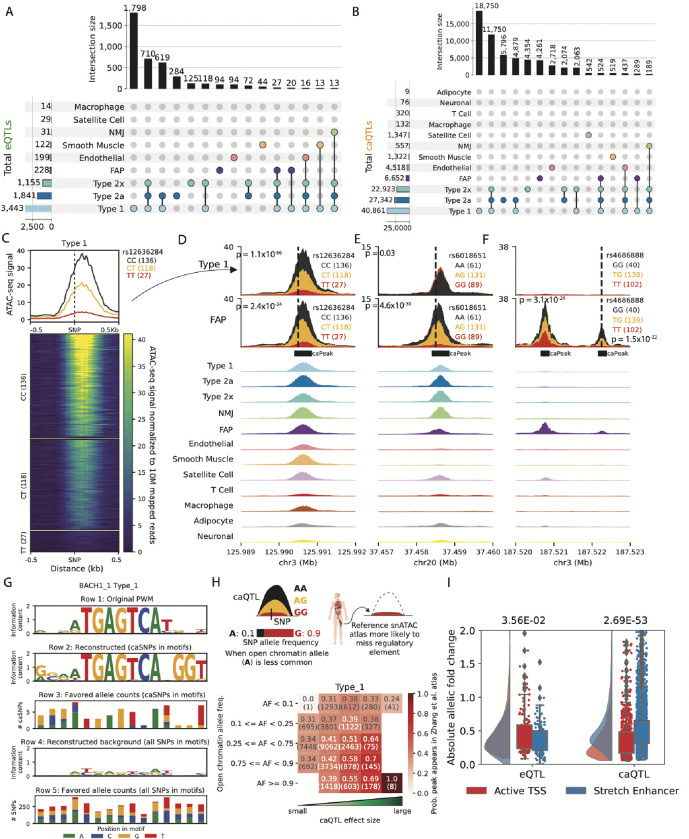
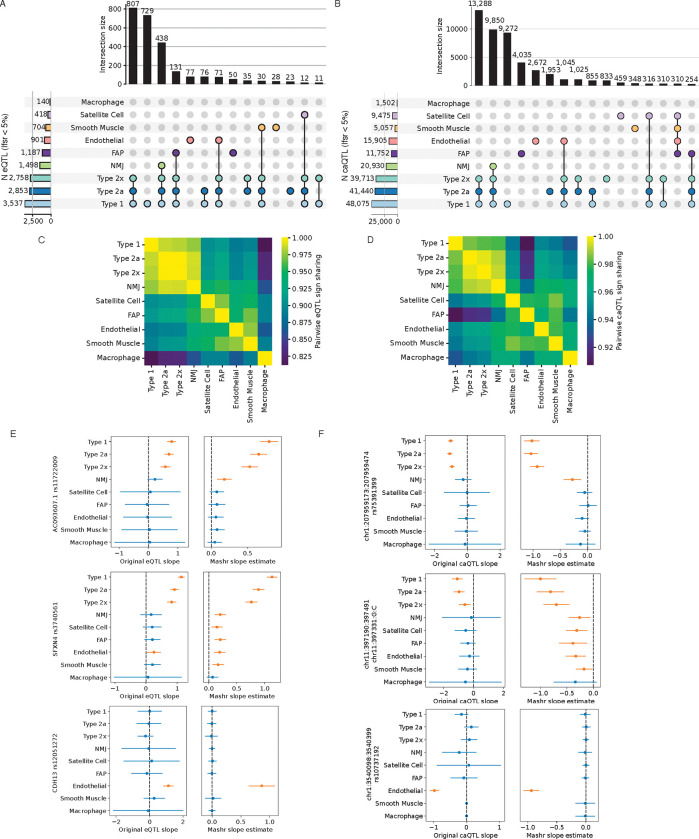
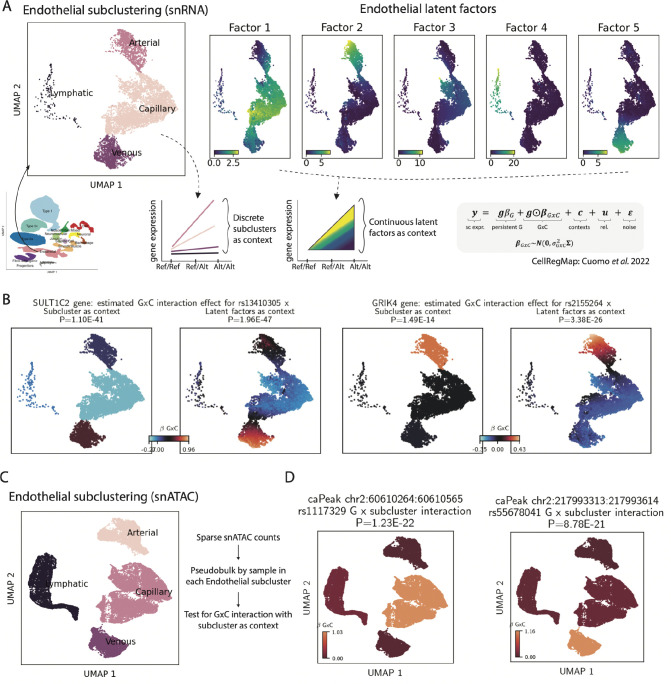
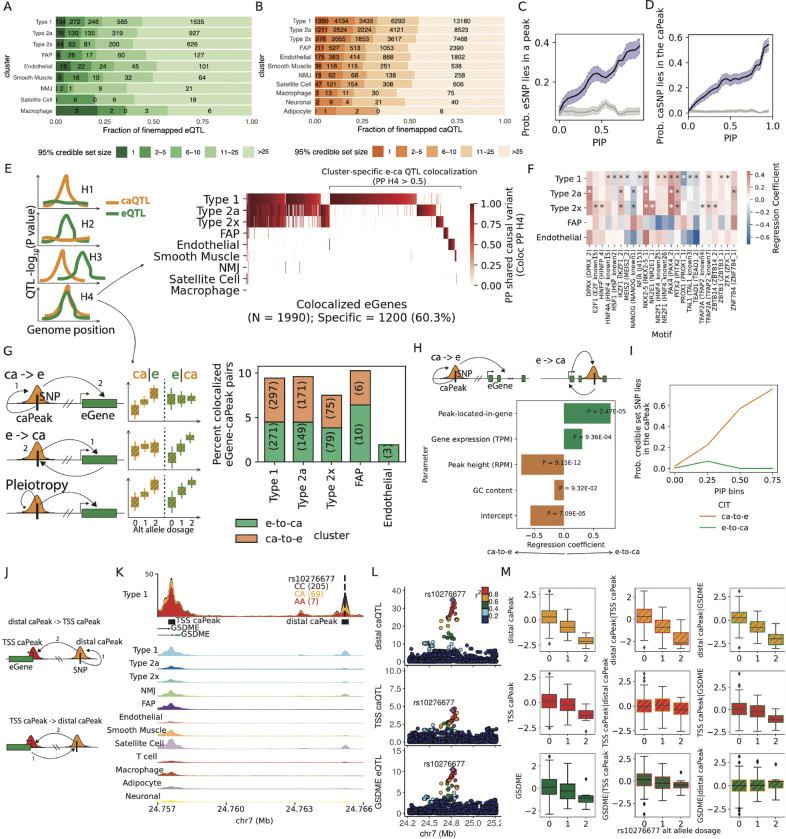
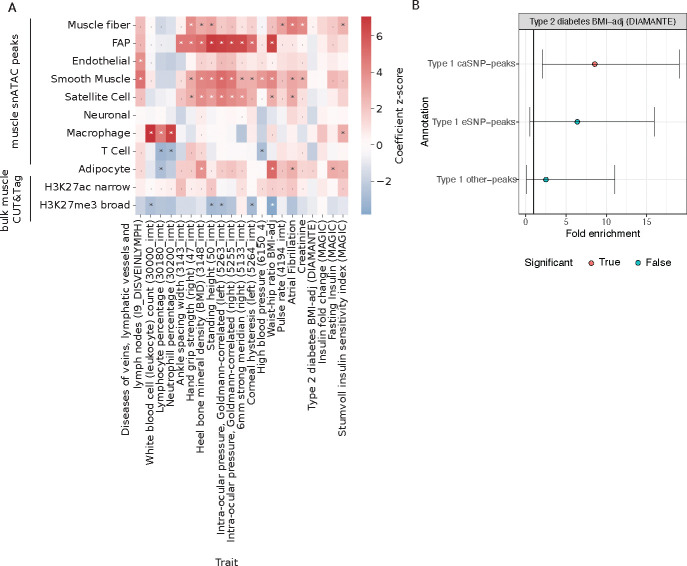
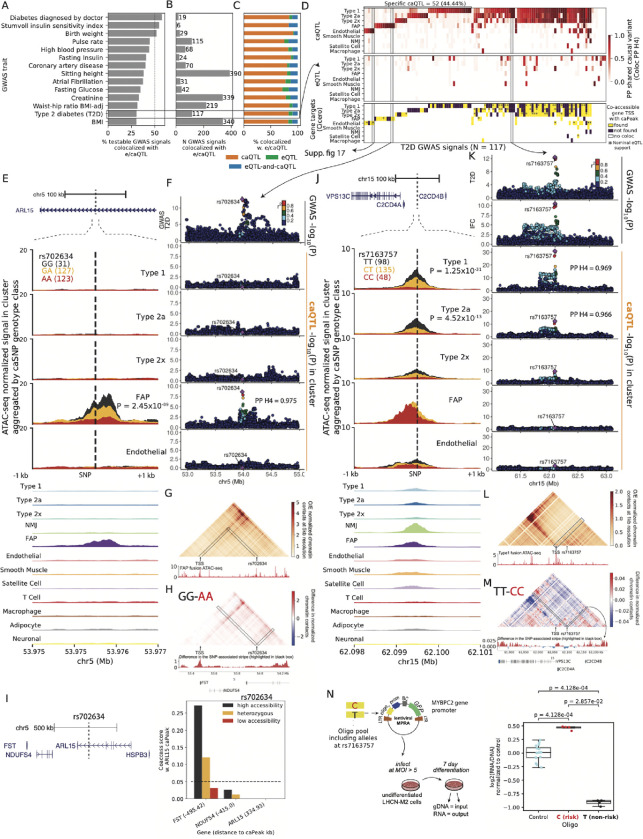
Skeletal muscle, the largest human organ by weight, is relevant in several polygenic metabolic traits and diseases including type 2 diabetes (T2D). Identifying genetic mechanisms underlying these traits requires pinpointing cell types, regulatory elements, target genes, and causal variants. Here, we use genetic multiplexing to generate population-scale single nucleus (sn) chromatin accessibility (snATAC-seq) and transcriptome (snRNA-seq) maps across 287 frozen human skeletal muscle biopsies representing nearly half a million nuclei. We identify 13 cell types and integrate genetic variation to discover >7,000 expression quantitative trait loci (eQTL) and >100,000 chromatin accessibility QTLs (caQTL) across cell types. Learning patterns of e/caQTL sharing across cell types increased precision of effect estimates. We identify high-resolution cell-states and context-specific e/caQTL with significant genotype by context interaction. We identify nearly 2,000 eGenes colocalized with caQTL and construct causal directional maps for chromatin accessibility and gene expression. Almost 3,500 genome-wide association study (GWAS) signals across 38 relevant traits colocalize with sn-e/caQTL, most in a cell-specific manner. These signals typically colocalize with caQTL and not eQTL, highlighting the importance of population-scale chromatin profiling for GWAS functional studies. Finally, our GWAS-caQTL colocalization data reveal distinct cell-specific regulatory paradigms. Our results illuminate the genetic regulatory architecture of human skeletal muscle at high resolution epigenomic, transcriptomic, and cell-state scales and serve as a template for population-scale multi-omic mapping in complex tissues and traits.
Conflict of interest statement
4.32Competing interests SCJP has a research grant from Pfizer.
Figures
Similar articles
-
Single cell variant to enhancer to gene map for coronary artery disease.medRxiv [Preprint]. 2024 Nov 13:2024.11.13.24317257. doi: 10.1101/2024.11.13.24317257. medRxiv. 2024. PMID: 39606421 Free PMC article. Preprint.
-
Impact of disease-associated chromatin accessibility QTLs across immune cell types and contexts.medRxiv [Preprint]. 2024 Dec 12:2024.12.05.24318552. doi: 10.1101/2024.12.05.24318552. medRxiv. 2024. PMID: 39711700 Free PMC article. Preprint.
-
Haplotype function score improves biological interpretation and cross-ancestry polygenic prediction of human complex traits.Elife. 2024 Apr 19;12:RP92574. doi: 10.7554/eLife.92574. Elife. 2024. PMID: 38639992 Free PMC article.
-
The effectiveness of abstinence-based and harm reduction-based interventions in reducing problematic substance use in adults who are experiencing homelessness in high income countries: A systematic review and meta-analysis: A systematic review.Campbell Syst Rev. 2024 Apr 21;20(2):e1396. doi: 10.1002/cl2.1396. eCollection 2024 Jun. Campbell Syst Rev. 2024. PMID: 38645303 Free PMC article. Review.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
Cited by
-
Effects of PreOperative radiotherapy in a preclinical glioblastoma model: a paradigm-shift approach.J Neurooncol. 2024 Sep;169(3):633-646. doi: 10.1007/s11060-024-04765-5. Epub 2024 Jul 22. J Neurooncol. 2024. PMID: 39037687
References
-
- Frontera W. R. & Ochala J. Skeletal Muscle: A Brief Review of Structure and Function. en. Calcified Tissue International 96, 183–195 (Mar. 2015). - PubMed
-
- Musi N. et al. Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects With Type 2 Diabetes. Diabetes 51, 2074–2081 (July 2002). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources