

The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies
- PMID: 38139154
- PMCID: PMC10743660
- DOI: 10.3390/ijms242417325
The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies
Abstract
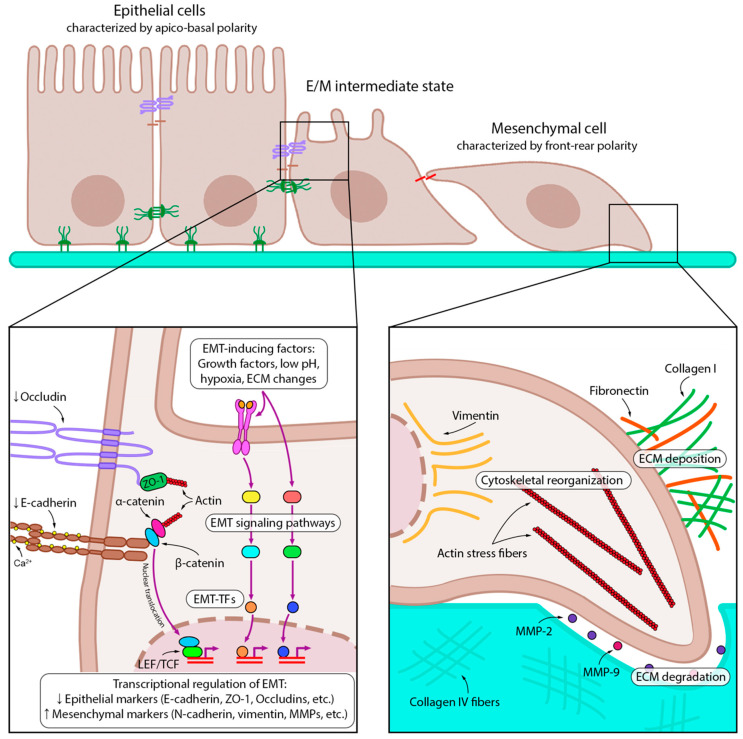
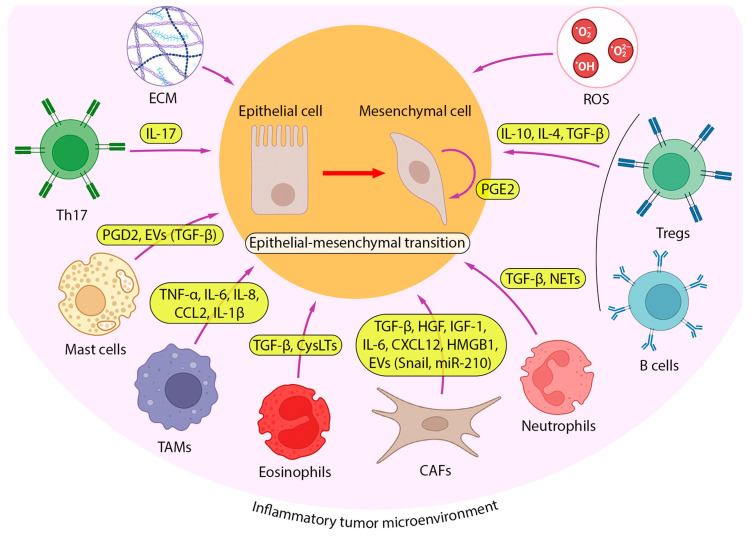
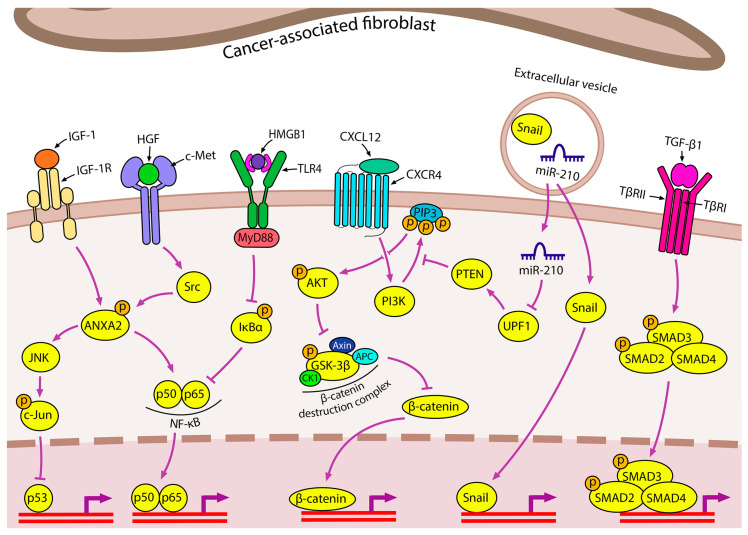
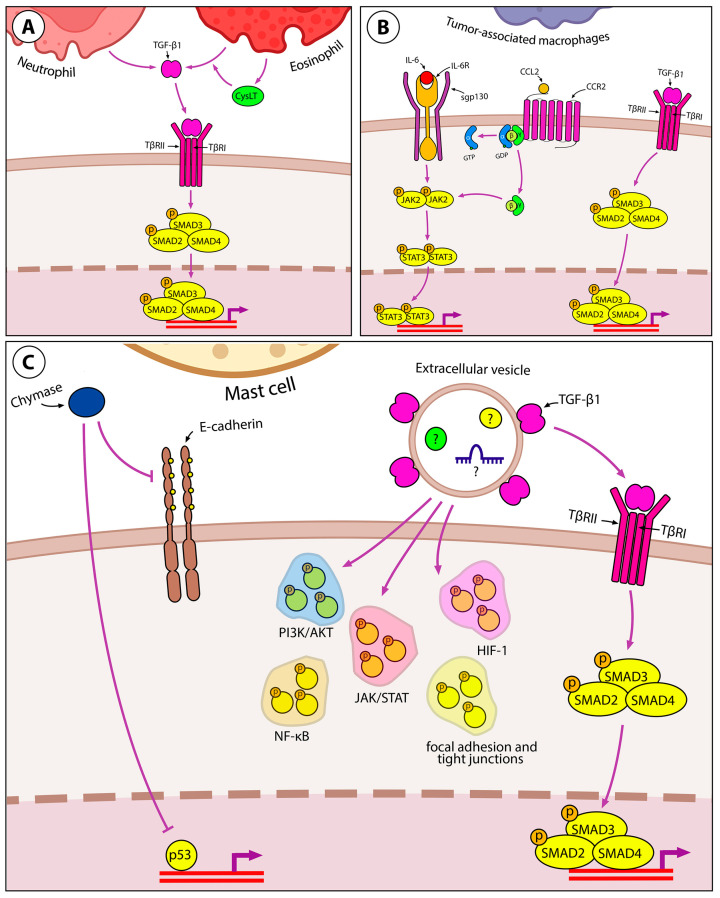
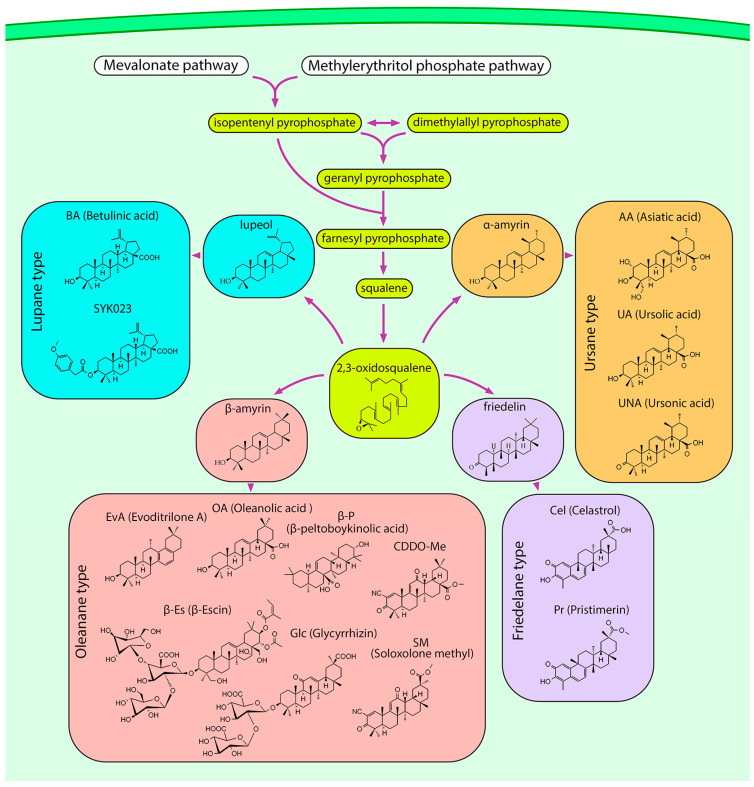
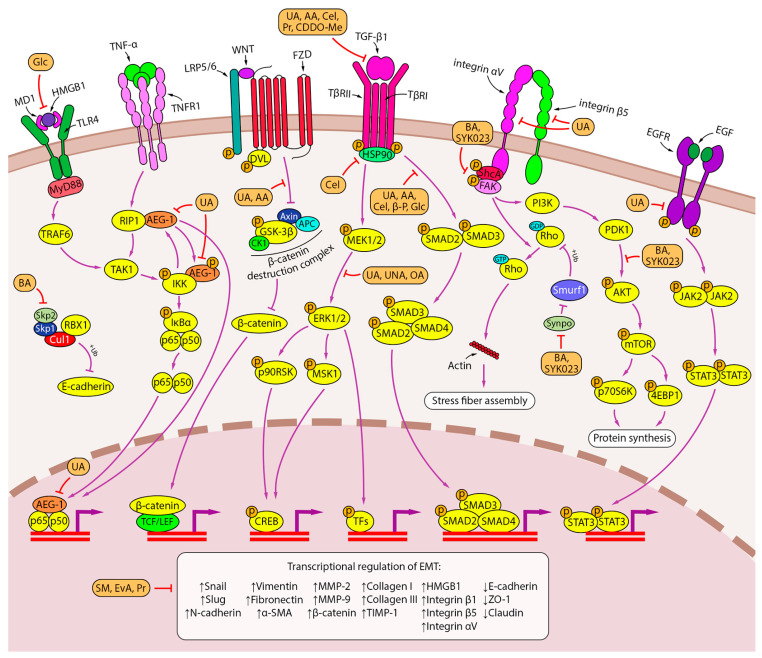
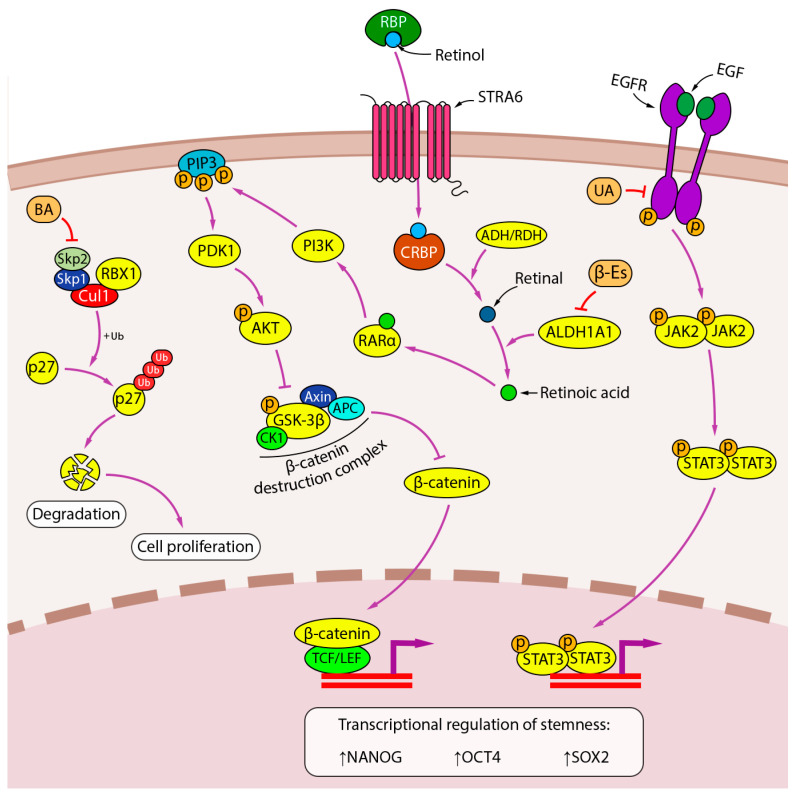
Lung cancer is the leading cause of cancer-related death worldwide. Its high mortality is partly due to chronic inflammation that accompanies the disease and stimulates cancer progression. In this review, we analyzed recent studies and highlighted the role of the epithelial-mesenchymal transition (EMT) as a link between inflammation and lung cancer. In the inflammatory tumor microenvironment (iTME), fibroblasts, macrophages, granulocytes, and lymphocytes produce inflammatory mediators, some of which can induce EMT. This leads to increased invasiveness of tumor cells and self-renewal of cancer stem cells (CSCs), which are associated with metastasis and tumor recurrence, respectively. Based on published data, we propose that inflammation-induced EMT may be a potential therapeutic target for the treatment of lung cancer. This prospect is partially realized in the development of EMT inhibitors based on pentacyclic triterpenoids (PTs), described in the second part of our study. PTs reduce the metastatic potential and stemness of tumor cells, making PTs promising candidates for lung cancer therapy. We emphasize that the high diversity of molecular mechanisms underlying inflammation-induced EMT far exceeds those that have been implicated in drug development. Therefore, analysis of information on the relationship between the iTME and EMT is of great interest and may provide ideas for novel treatment approaches for lung cancer.
Keywords: aggressiveness; epithelial-to-mesenchymal transition; inflammation; mechanism of action; natural products; pulmonary malignancy; tumor stem cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The Role of Cancer Stem Cells in Recurrent and Drug-Resistant Lung Cancer.Adv Exp Med Biol. 2016;890:57-74. doi: 10.1007/978-3-319-24932-2_4. Adv Exp Med Biol. 2016. PMID: 26703799 Review.
-
Epithelial Mesenchymal Transition in Aggressive Lung Cancers.Adv Exp Med Biol. 2016;890:37-56. doi: 10.1007/978-3-319-24932-2_3. Adv Exp Med Biol. 2016. PMID: 26703798 Review.
-
Emodin reduces Breast Cancer Lung Metastasis by suppressing Macrophage-induced Breast Cancer Cell Epithelial-mesenchymal transition and Cancer Stem Cell formation.Theranostics. 2020 Jul 9;10(18):8365-8381. doi: 10.7150/thno.45395. eCollection 2020. Theranostics. 2020. PMID: 32724475 Free PMC article.
-
Cyano Enone-Bearing Triterpenoid Soloxolone Methyl Inhibits Epithelial-Mesenchymal Transition of Human Lung Adenocarcinoma Cells In Vitro and Metastasis of Murine Melanoma In Vivo.Molecules. 2020 Dec 14;25(24):5925. doi: 10.3390/molecules25245925. Molecules. 2020. PMID: 33327637 Free PMC article.
-
Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets.Stem Cells. 2016 Aug;34(8):1997-2007. doi: 10.1002/stem.2406. Epub 2016 Jun 20. Stem Cells. 2016. PMID: 27251010 Review.
Cited by
-
Morphine promotes non-small cell lung cancer progression by downregulating E-cadherin via the PI3K/AKT/mTOR pathway.Sci Rep. 2024 Sep 10;14(1):21130. doi: 10.1038/s41598-024-72198-1. Sci Rep. 2024. PMID: 39256509 Free PMC article.
-
Pulmonary mucinous adenocarcinoma: An overview of pathophysiology and advancements in treatment.Heliyon. 2024 Mar 29;10(9):e28881. doi: 10.1016/j.heliyon.2024.e28881. eCollection 2024 May 15. Heliyon. 2024. PMID: 38694119 Free PMC article. Review.
References
-
- Arseniev A.I., Nefedov A.O., Novikov S.N., Barchuk A.A., Tarkov S.A., Kostitsin K.A., Nefedova A.V., Aristidov N.Y., Semiletova Y.V., Ryazankina A.A. Algorithms of non-invasive, minimally-invasive and invasive diagnostics for lung cancer (review) Prev. Clin. Med. 2021;2:69–77. (In Russian)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
