

The landscape of genomic structural variation in Indigenous Australians
- PMID: 38093003
- PMCID: PMC10733147
- DOI: 10.1038/s41586-023-06842-7
The landscape of genomic structural variation in Indigenous Australians
Abstract
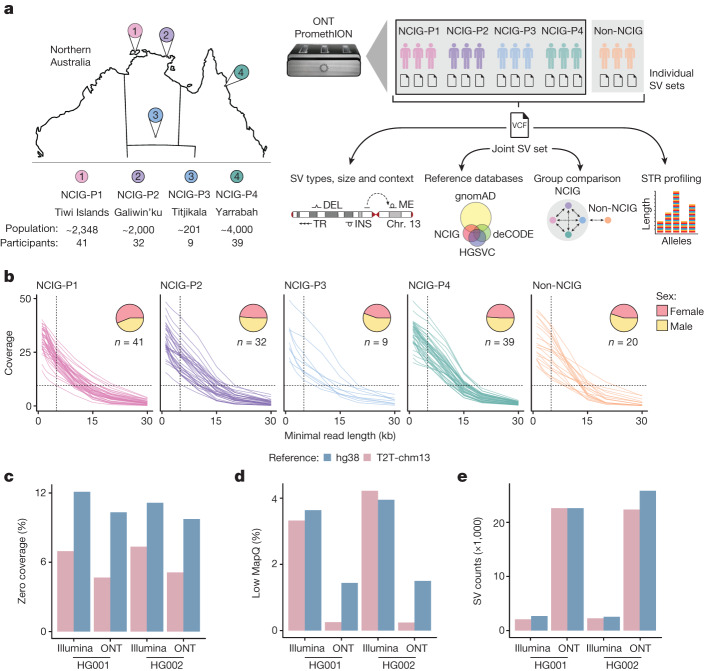
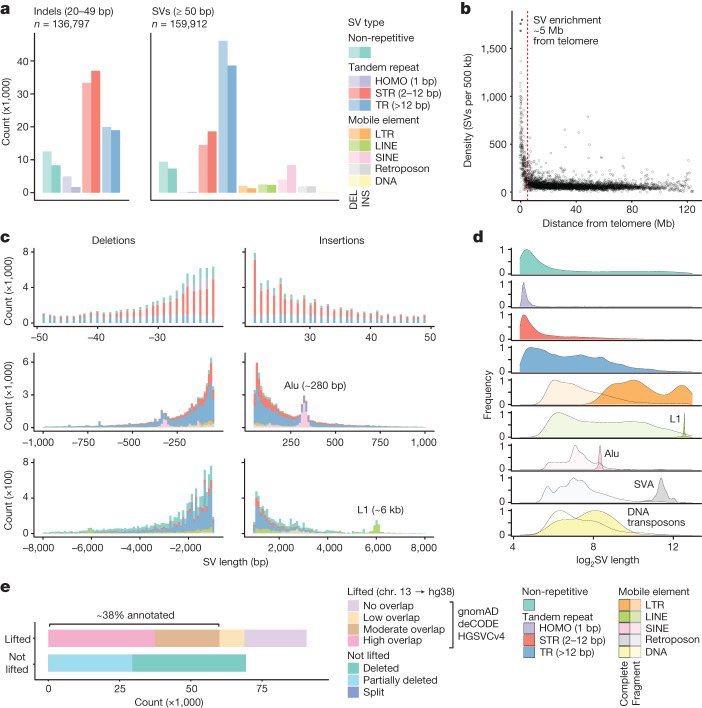
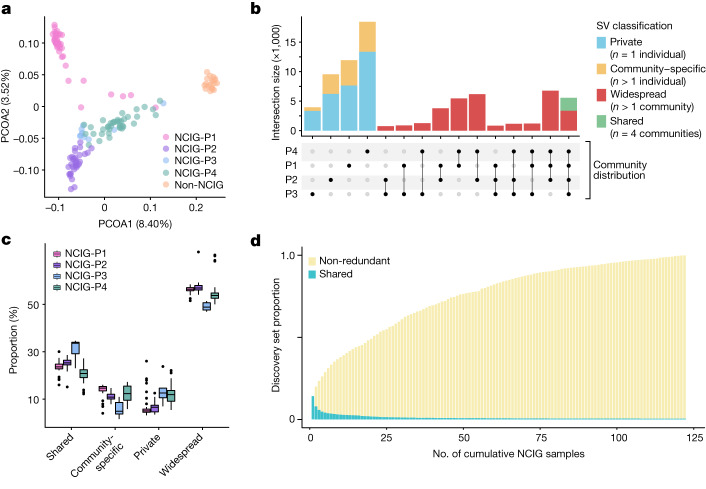
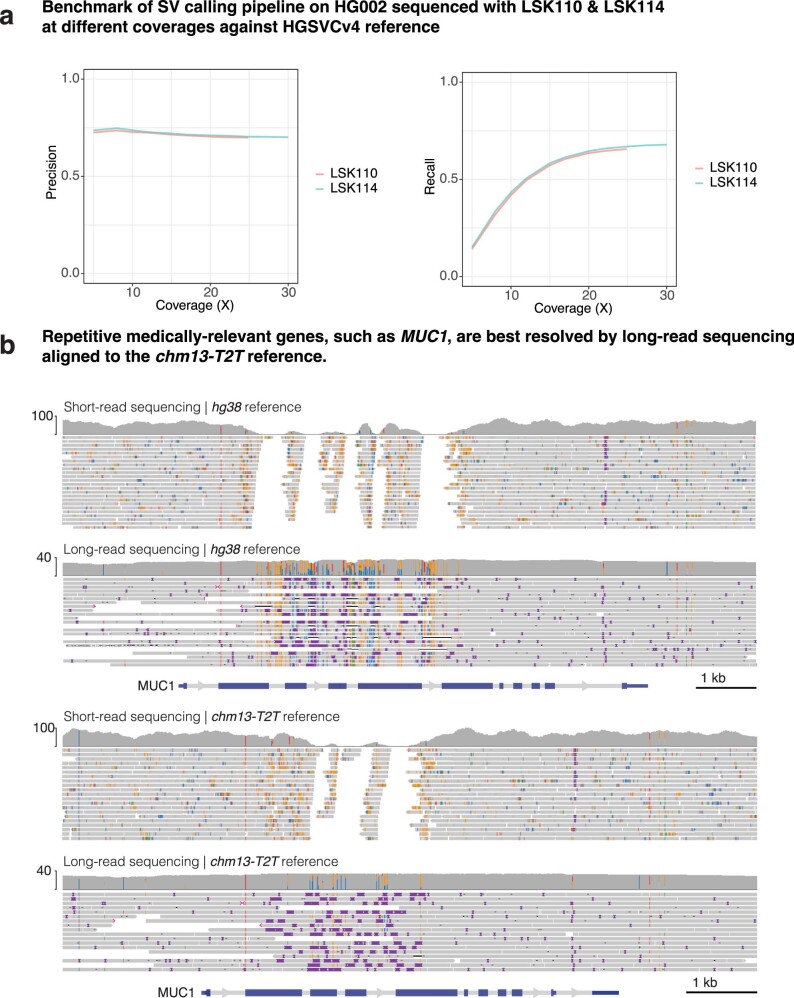
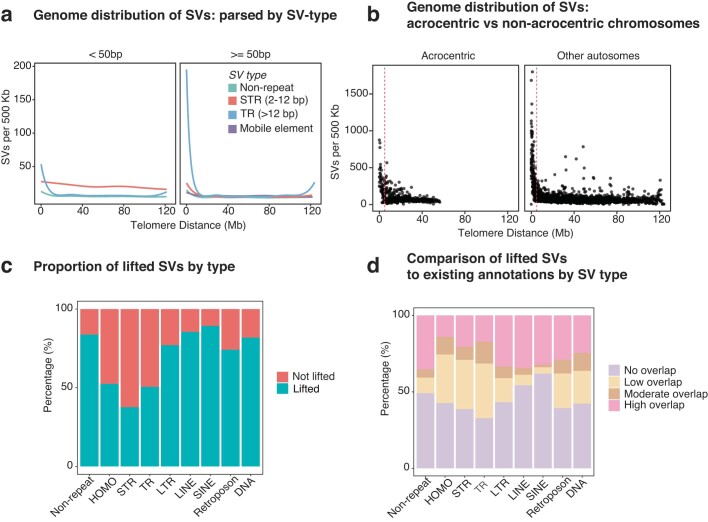
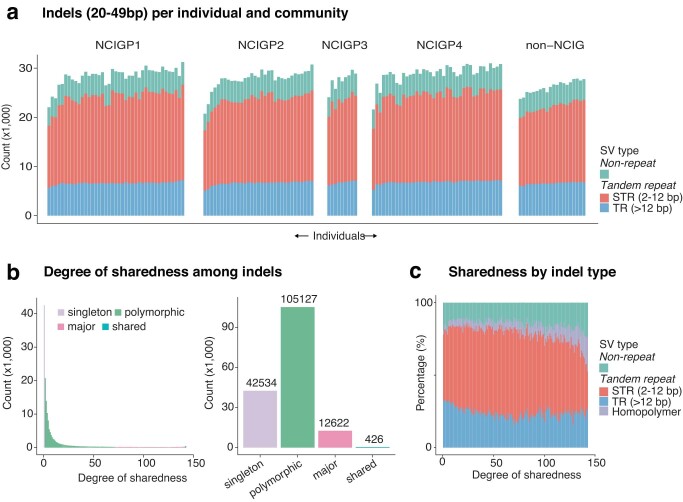
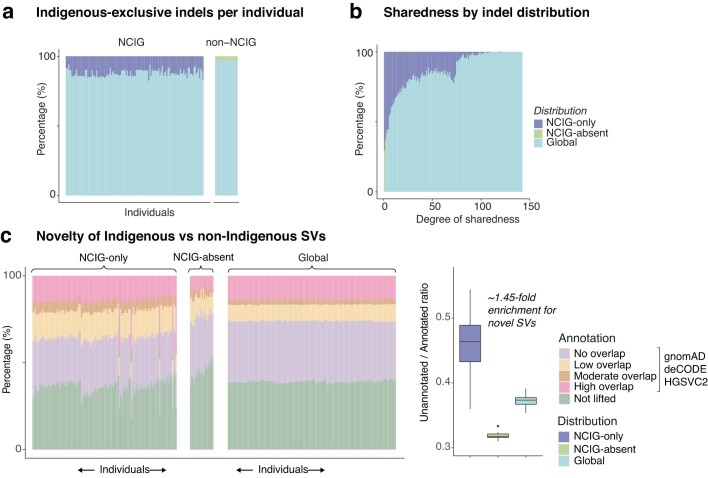
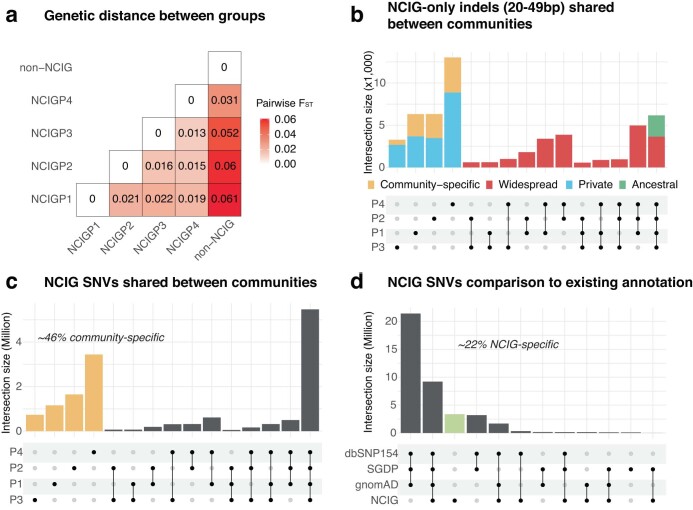
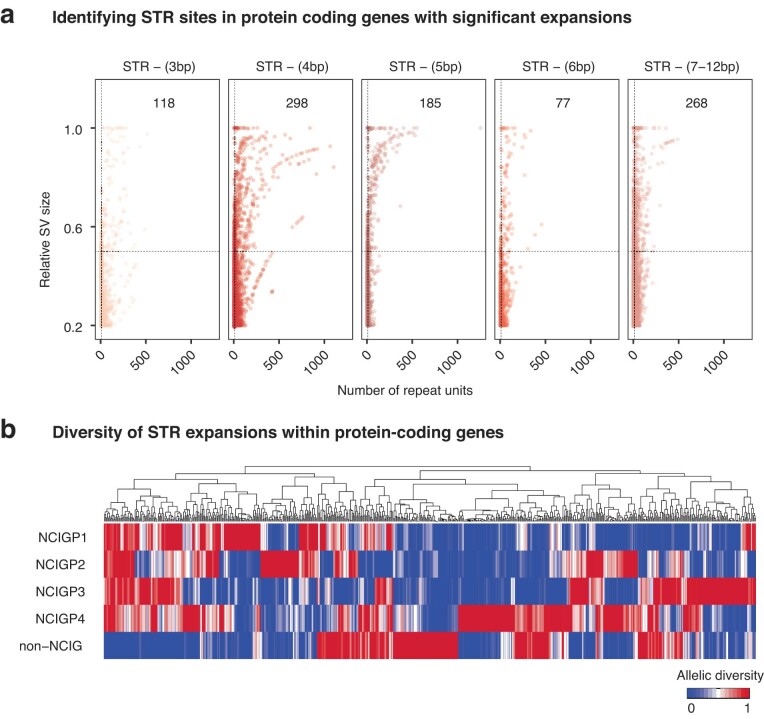
Indigenous Australians harbour rich and unique genomic diversity. However, Aboriginal and Torres Strait Islander ancestries are historically under-represented in genomics research and almost completely missing from reference datasets1-3. Addressing this representation gap is critical, both to advance our understanding of global human genomic diversity and as a prerequisite for ensuring equitable outcomes in genomic medicine. Here we apply population-scale whole-genome long-read sequencing4 to profile genomic structural variation across four remote Indigenous communities. We uncover an abundance of large insertion-deletion variants (20-49 bp; n = 136,797), structural variants (50 b-50 kb; n = 159,912) and regions of variable copy number (>50 kb; n = 156). The majority of variants are composed of tandem repeat or interspersed mobile element sequences (up to 90%) and have not been previously annotated (up to 62%). A large fraction of structural variants appear to be exclusive to Indigenous Australians (12% lower-bound estimate) and most of these are found in only a single community, underscoring the need for broad and deep sampling to achieve a comprehensive catalogue of genomic structural variation across the Australian continent. Finally, we explore short tandem repeats throughout the genome to characterize allelic diversity at 50 known disease loci5, uncover hundreds of novel repeat expansion sites within protein-coding genes, and identify unique patterns of diversity and constraint among short tandem repeat sequences. Our study sheds new light on the dimensions and dynamics of genomic structural variation within and beyond Australia.
© 2023. The Author(s).
Conflict of interest statement
This project receives partial in-kind support from Oxford Nanopore Technologies (ONT) under an ongoing collaboration agreement. A.L.M.R., J.M.H., H.G., H.R.P. and I.W.D. have previously received travel and accommodation expenses from ONT to speak at conferences. H.G. and I.W.D. have paid consultant roles with Sequin Pty Ltd. O.M.S. and A.W.H. hold equity in Seonix Pty Ltd. The authors declare no other competing interests.
Figures







Similar articles
-
Australian Indigenous genomes are highly diverse and unlike those anywhere else.Nature. 2024 Jan;625(7993):15-16. doi: 10.1038/d41586-023-04006-1. Nature. 2024. PMID: 38093071 No abstract available.
-
Precision medicine in Australia: indigenous health professionals are needed to improve equity for Aboriginal and Torres Strait Islanders.Int J Equity Health. 2024 Jul 4;23(1):134. doi: 10.1186/s12939-024-02202-7. Int J Equity Health. 2024. PMID: 38965527 Free PMC article.
-
Indigenous Australian genomes show deep structure and rich novel variation.Nature. 2023 Dec;624(7992):593-601. doi: 10.1038/s41586-023-06831-w. Epub 2023 Dec 13. Nature. 2023. PMID: 38093005 Free PMC article.
-
Towards precision cancer medicine for Aboriginal and Torres Strait Islander cancer health equity.Med J Aust. 2024 Jul 1;221(1):68-73. doi: 10.5694/mja2.52346. Med J Aust. 2024. PMID: 38946636 Review.
-
Measuring what counts in Aboriginal and Torres Strait Islander care: a review of general practice datasets available for assessing chronic disease care.Aust J Prim Health. 2024 Jul;30:PY24017. doi: 10.1071/PY24017. Aust J Prim Health. 2024. PMID: 38981000 Review.
Cited by
-
A call to action to scale up research and clinical genomic data sharing.Nat Rev Genet. 2025 Feb;26(2):141-147. doi: 10.1038/s41576-024-00776-0. Epub 2024 Oct 7. Nat Rev Genet. 2025. PMID: 39375561 Review.
-
An assessment of the genomic structural variation landscape in Sub-Saharan African populations.Res Sq [Preprint]. 2024 Jul 8:rs.3.rs-4485126. doi: 10.21203/rs.3.rs-4485126/v1. Res Sq. 2024. PMID: 39041024 Free PMC article. Preprint.
-
Australian Indigenous genomes are highly diverse and unlike those anywhere else.Nature. 2024 Jan;625(7993):15-16. doi: 10.1038/d41586-023-04006-1. Nature. 2024. PMID: 38093071 No abstract available.
-
Precision medicine in Australia: indigenous health professionals are needed to improve equity for Aboriginal and Torres Strait Islanders.Int J Equity Health. 2024 Jul 4;23(1):134. doi: 10.1186/s12939-024-02202-7. Int J Equity Health. 2024. PMID: 38965527 Free PMC article.
-
Increasing Diversity, Equity, Inclusion, and Accessibility in Rare Disease Clinical Trials.Pharmaceut Med. 2024 Jul;38(4):261-276. doi: 10.1007/s40290-024-00529-8. Epub 2024 Jul 9. Pharmaceut Med. 2024. PMID: 38977611 Review.
References
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
