

Integrated DNA Methylation and Transcriptomics Analyses of Lacrimal Glands Identify the Potential Genes Implicated in the Development of Sjögren's Syndrome-Related Dry Eye
- PMID: 38050559
- PMCID: PMC10693829
- DOI: 10.2147/JIR.S440263
Integrated DNA Methylation and Transcriptomics Analyses of Lacrimal Glands Identify the Potential Genes Implicated in the Development of Sjögren's Syndrome-Related Dry Eye
Abstract
Purpose: Sjögren's syndrome-related dry eye (SS-related dry eye) is an intractable autoimmune disease characterized by chronic inflammation of lacrimal glands (LGs), where epigenetic factors are proven to play a crucial role in the pathogenesis of this disease. However, the alteration of DNA methylation in LGs and its role in the pathogenesis of SS-related dry eye is still unknown. Here, we performed an integrated analysis of DNA methylation and RNA-Seq data in LGs to identify novel DNA methylation-regulated differentially expressed genes (MeDEGs) in the pathogenesis of SS-related dry eye.
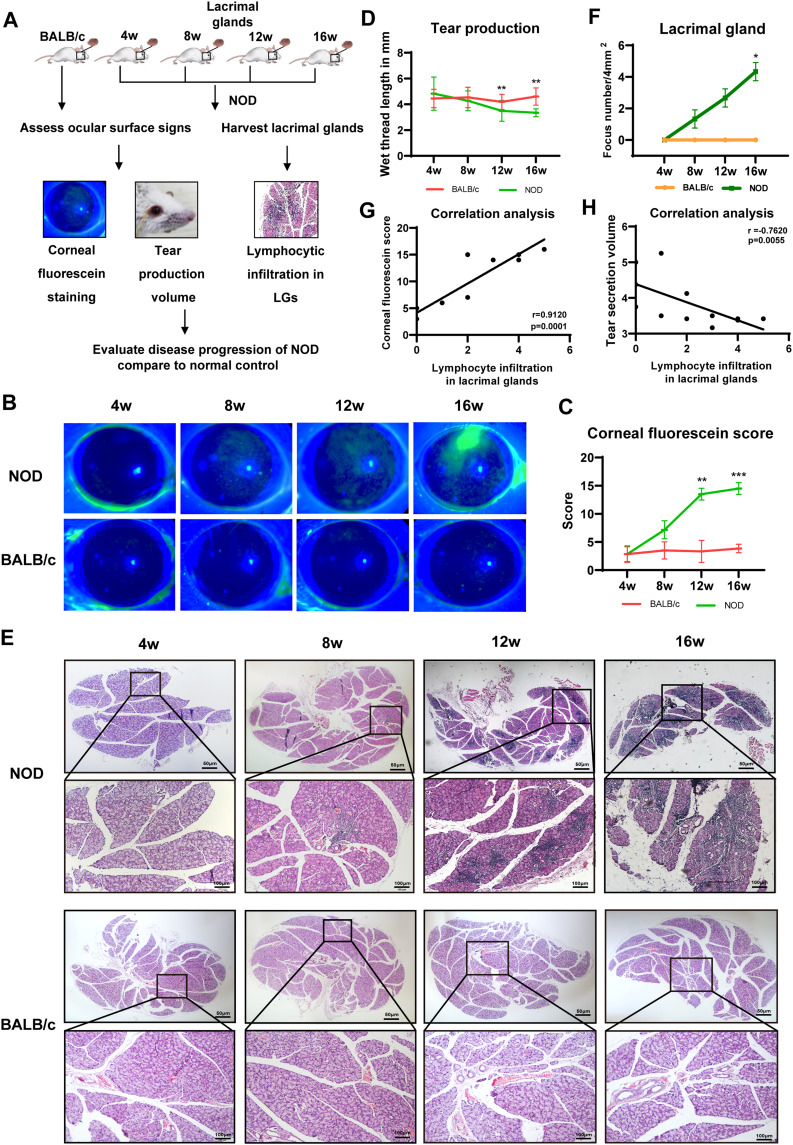
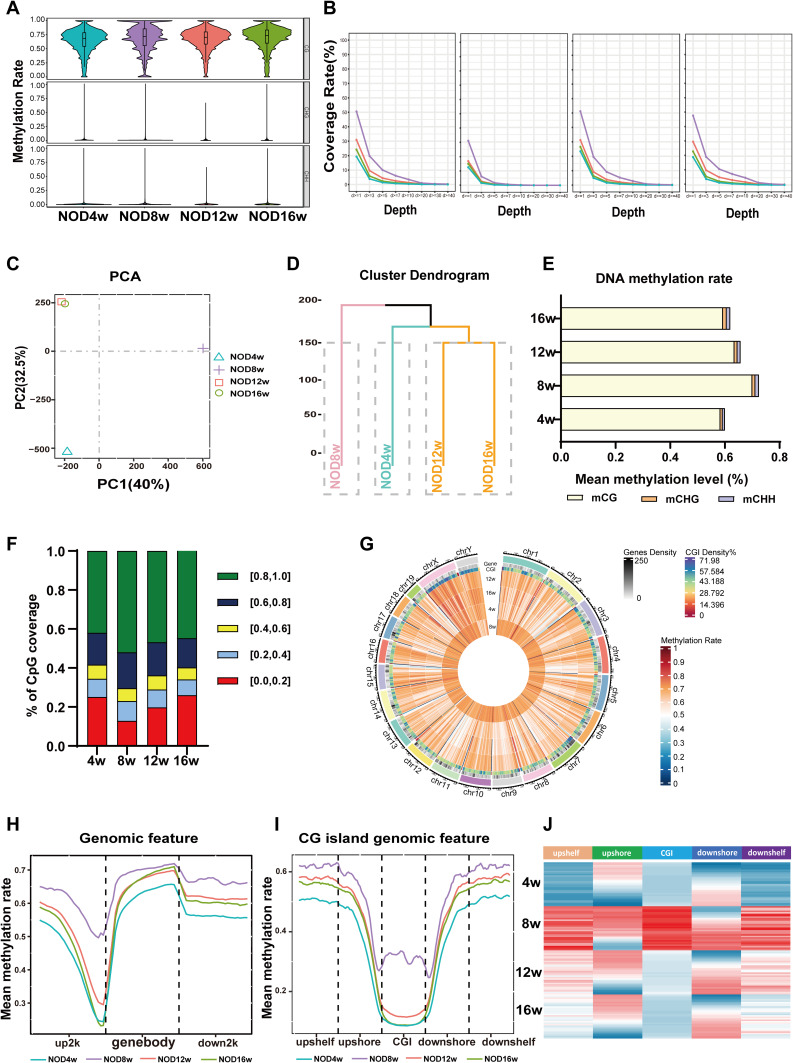
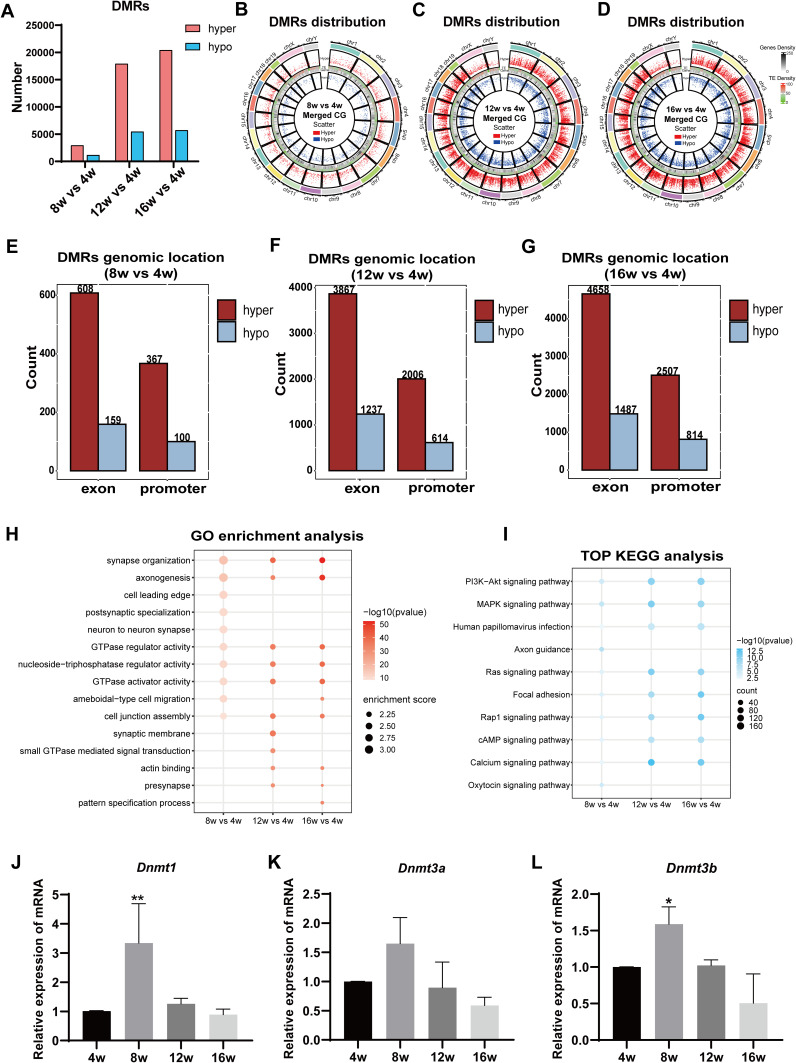
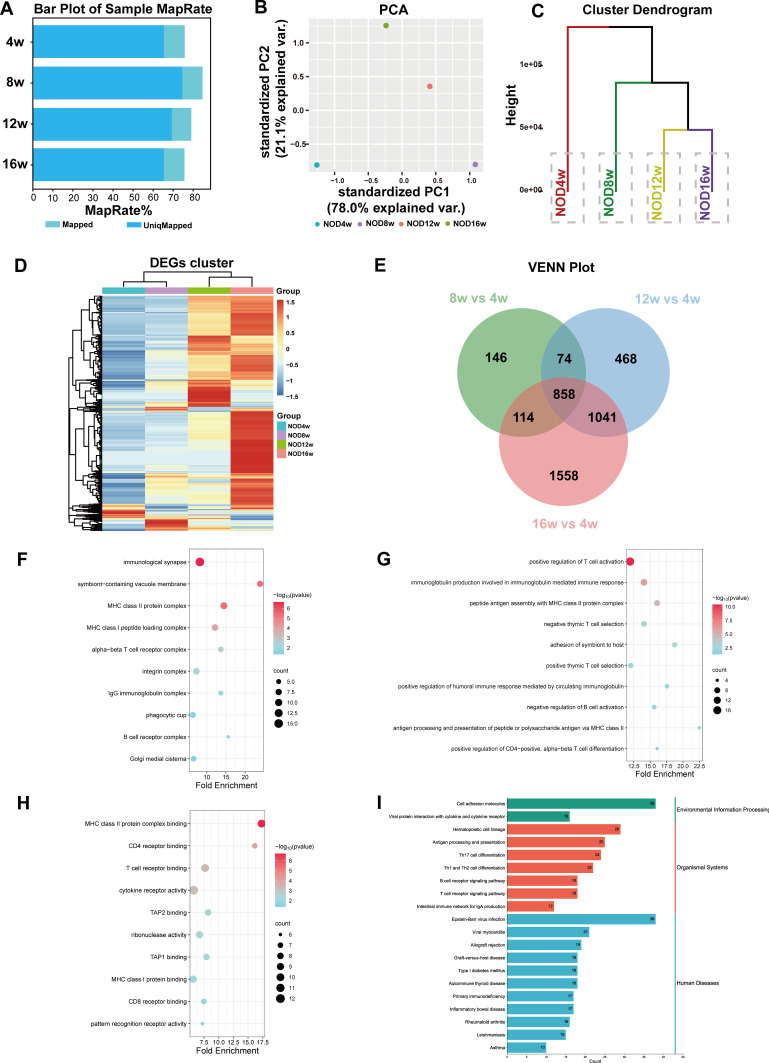
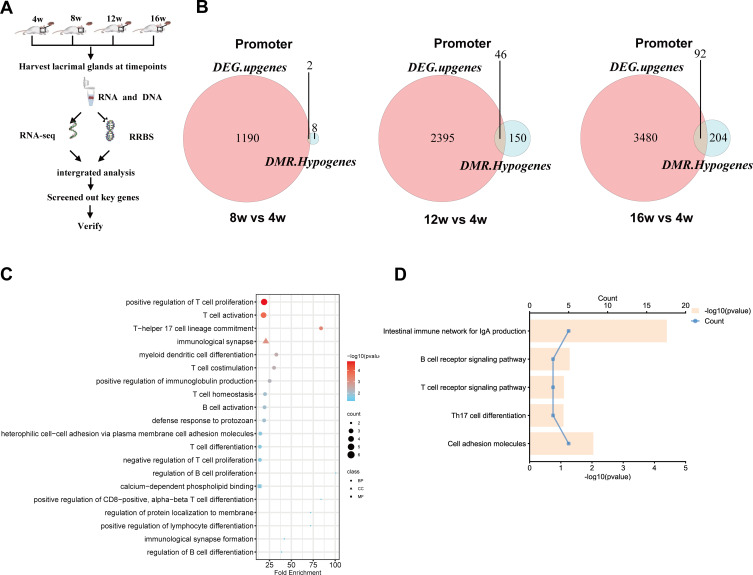
Methods: The DNA methylation and transcription profiles of LGs in NOD mice at different stages of SS-related dry eye (4-, 8-, 12- and 16 weeks old) were generated by reduced representation bisulfite sequencing (RRBS) and RNA-Seq. The differentially methylated genes (DMGs) and differentially expressed genes (DEGs) were analyzed by MethylKit R package and edgeR. Correlation analysis between methylation level and mRNA expression was conducted with R software. The functional correlation of DMGs and DEGs was analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Finally, LG tissues from another litter of NOD mice were collected for methylation-specific polymerase chain reaction (MSP) and quantitative real-time PCR (qRT-PCR) to validate the methylation and expression levels of key genes. CD4+ cell infiltration of LGs was detected by immunofluorescence staining.
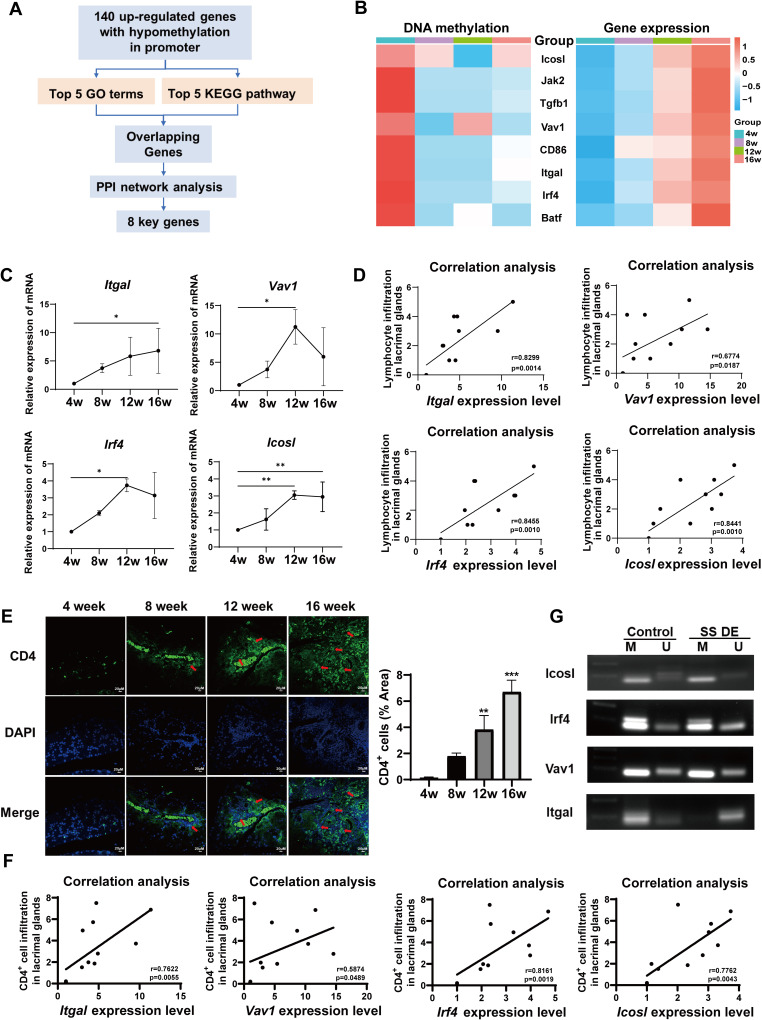
Results: Hypermethylation of LGs was identified in NOD mice with the progression of SS-related dry eye and the DMGs were mainly enriched in the GTPases activation and Ras signaling pathway. RNA-seq analysis revealed 1321, 2549, and 3712 DEGs in the 8-, 12- and 16-week-old NOD mice compared with 4-week-old normal control mice. For GO analysis, the DEGs were mainly enriched in T cell immune responses. Further, a total of 140 MeDEGs were obtained by integrated analysis of methylome and transcriptome, which were primarily enriched in T cell activation, proliferation and differentiation. Based on the main GO terms and KEGG pathways of MeDEGs, 8 genes were screened out. The expression levels of these key genes, especially Itgal, Vav1, Irf4 and Icosl, were verified to elevate after the onset of SS-related dry eye in NOD mice and positively correlated with the extent of inflammatory cell infiltration in LGs. Immunofluorescence assay revealed that CD4+ cell infiltration dramatically increased in LGs of SS-related dry eye mice compared with the control mice. And the expression levels of four genes showed significantly positive correlation with the extent of CD4+ cell infiltration in LGs. MSP showed the hypomethylation of the Irf4 and Itgal promoters in NOD mice with SS-related dry eye compared to control group.
Conclusion: Our study revealed the critical role of epigenetic regulation of T cell immunity-related genes in the progression of SS-related dry eye and reminded us that DNA methylation-regulated genes such as Itgal, Vav1, Irf4 and Icosl may be used as new targets for SS-related dry eye therapy.
Keywords: DNA methylation; RNA-seq; Sjögren’s syndrome-related dry eye; T cell-mediated immune response; lacrimal gland.
© 2023 Sun et al.
Conflict of interest statement
The authors reported no conflicts of interest in this work.
Figures
Similar articles
-
Exploration of the pathogenesis of Sjögren's syndrome via DNA methylation and transcriptome analyses.Clin Rheumatol. 2022 Sep;41(9):2765-2777. doi: 10.1007/s10067-022-06200-4. Epub 2022 May 13. Clin Rheumatol. 2022. PMID: 35562622
-
Identification of DNA methylation-regulated differentially expressed genes in RA by integrated analysis of DNA methylation and RNA-Seq data.J Transl Med. 2022 Oct 22;20(1):481. doi: 10.1186/s12967-022-03664-5. J Transl Med. 2022. PMID: 36273177 Free PMC article.
-
Arginase 1 is involved in lacrimal hyposecretion in male NOD mice, a model of Sjögren's syndrome, regardless of dacryoadenitis status.J Physiol. 2020 Nov;598(21):4907-4925. doi: 10.1113/JP280090. Epub 2020 Aug 29. J Physiol. 2020. PMID: 32780506 Free PMC article.
-
Review of genes potentially related to hyposecretion in male non-obese diabetic (NOD) mice, a Sjögren's syndrome model.J Oral Biosci. 2023 Sep;65(3):211-217. doi: 10.1016/j.job.2023.05.001. Epub 2023 May 18. J Oral Biosci. 2023. PMID: 37209839 Review.
-
Autoimmune Epithelitis and Chronic Inflammation in Sjögren's Syndrome-Related Dry Eye Disease.Int J Mol Sci. 2021 Oct 30;22(21):11820. doi: 10.3390/ijms222111820. Int J Mol Sci. 2021. PMID: 34769250 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
