

Genome editing for sickle cell disease: still time to correct?
- PMID: 38027257
- PMCID: PMC10652763
- DOI: 10.3389/fped.2023.1249275
Genome editing for sickle cell disease: still time to correct?
Abstract
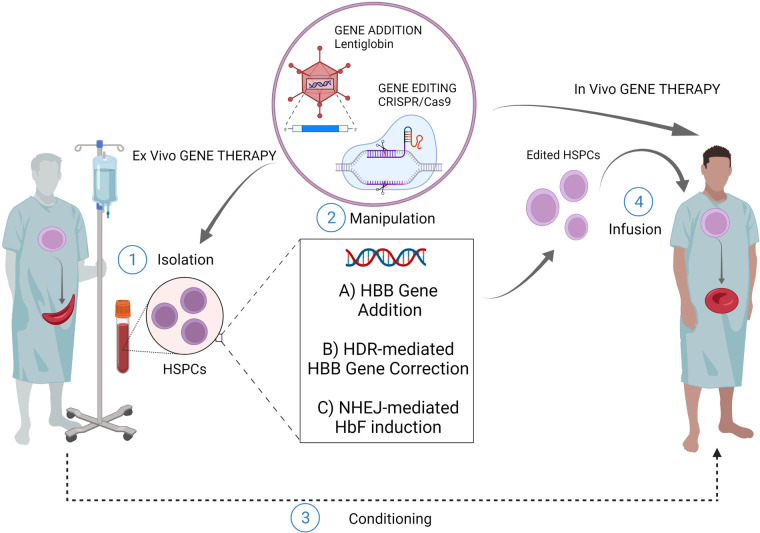
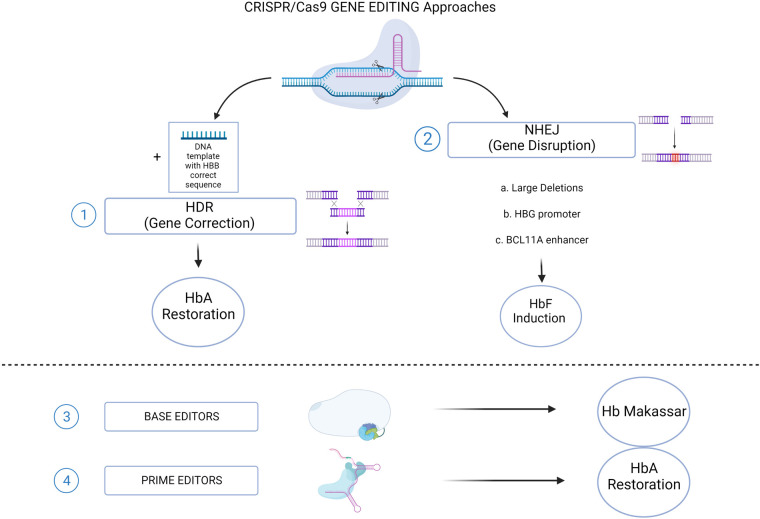
Sickle cell disease (SCD) is an inherited blood disorder, due to a single point mutation in the β-globin gene (HBB) leading to multisystemic manifestations and it affects millions of people worldwide. The monogenic nature of the disease and the availability of autologous hematopoietic stem cells (HSCs) make this disorder an ideal candidate for gene modification strategies. Notably, significant advances in the field of gene therapy and genome editing that took place in the last decade enabled the possibility to develop several strategies for the treatment of SCD. These curative approaches were firstly based on the correction of disease-causing mutations holding the promise for a specific, effective and safe option for patients. Specifically, gene-editing approaches exploiting the homology directed repair pathway were investigated, but soon their limited efficacy in quiescent HSC has curbed their wider development. On the other hand, a number of studies on globin gene regulation, led to the development of several genome editing strategies based on the reactivation of the fetal γ-globin gene (HBG) by nuclease-mediated targeting of HBG-repressor elements. Although the efficiency of these strategies seems to be confirmed in preclinical and clinical studies, very little is known about the long-term consequences of these modifications. Moreover, the potential genotoxicity of these nuclease-based strategies must be taken into account, especially when associated with high targeting rates. The recent introduction of nuclease-free genome editing technologies brought along the potential for safer strategies for SCD gene correction, which may also harbor significant advantages over HBG-reactivating ones. In this Review, we discuss the recent advances in genome editing strategies for the correction of SCD-causing mutations trying to recapitulate the promising strategies currently available and their relative strengths and weaknesses.
Keywords: CRISPR/Cas9; fetal hemoglobin reactivation; gene editing; gene therapy; globin genes regulation; sickle cell disease.
© 2023 Ceglie, Lecis, Canciani, Algeri and Frati.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Clinical genome editing to treat sickle cell disease-A brief update.Front Med (Lausanne). 2023 Jan 9;9:1065377. doi: 10.3389/fmed.2022.1065377. eCollection 2022. Front Med (Lausanne). 2023. PMID: 36698803 Free PMC article. Review.
-
Genome editing approaches to β-hemoglobinopathies.Prog Mol Biol Transl Sci. 2021;182:153-183. doi: 10.1016/bs.pmbts.2021.01.025. Epub 2021 Mar 1. Prog Mol Biol Transl Sci. 2021. PMID: 34175041
-
Hematopoietic-Stem-Cell-Targeted Gene-Addition and Gene-Editing Strategies for β-hemoglobinopathies.Cell Stem Cell. 2021 Feb 4;28(2):191-208. doi: 10.1016/j.stem.2021.01.001. Cell Stem Cell. 2021. PMID: 33545079 Review.
-
Combination of lentiviral and genome editing technologies for the treatment of sickle cell disease.Mol Ther. 2022 Jan 5;30(1):145-163. doi: 10.1016/j.ymthe.2021.08.019. Epub 2021 Aug 19. Mol Ther. 2022. PMID: 34418541 Free PMC article.
-
Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates.Mol Ther. 2019 Aug 7;27(8):1389-1406. doi: 10.1016/j.ymthe.2019.05.014. Epub 2019 May 24. Mol Ther. 2019. PMID: 31178391 Free PMC article.
Cited by
-
Construction and Stability of All-in-One Adenovirus Vectors Simultaneously Expressing Four and Eight Multiplex Guide RNAs and Cas9 Nickase.Int J Mol Sci. 2024 Aug 12;25(16):8783. doi: 10.3390/ijms25168783. Int J Mol Sci. 2024. PMID: 39201470 Free PMC article.
-
Regulatory Assessment of Casgevy for the Treatment of Transfusion-Dependent β-Thalassemia and Sickle Cell Disease with Recurrent Vaso-Occlusive Crises.Curr Issues Mol Biol. 2024 Jul 30;46(8):8209-8225. doi: 10.3390/cimb46080485. Curr Issues Mol Biol. 2024. PMID: 39194702 Free PMC article. Review.
References
-
- Kanter J, Walters MC, Hsieh MM, Krishnamurti L, Kwiatkowski J, Kamble RT, et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood. (2016) 128:1176. 10.1182/blood.V128.22.1176.1176 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources