

Protocol for transcriptome assembly by the TransBorrow algorithm
- PMID: 38023349
- PMCID: PMC10640700
- DOI: 10.1093/biomethods/bpad028
Protocol for transcriptome assembly by the TransBorrow algorithm
Abstract
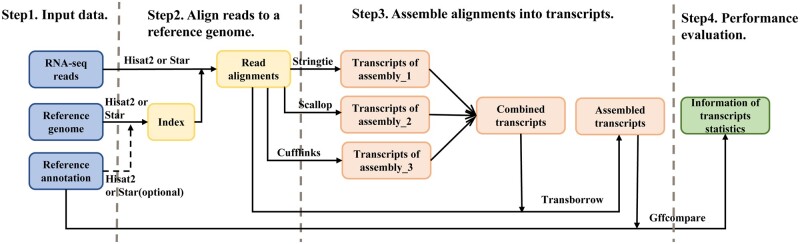
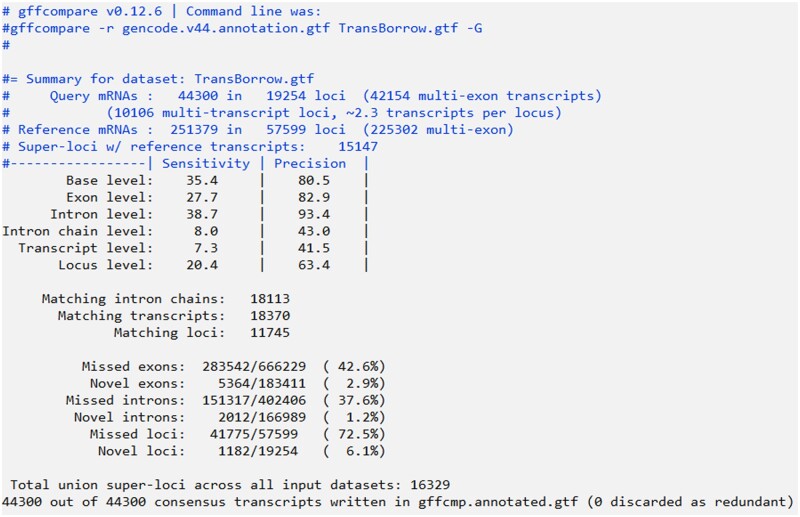
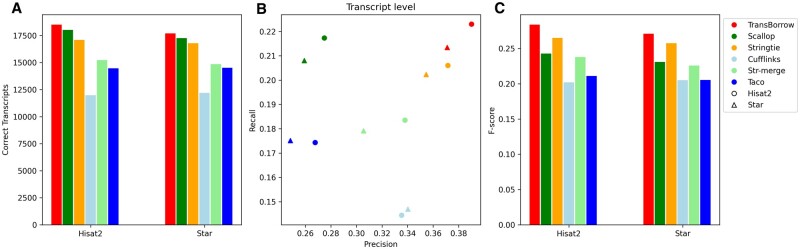
High-throughput RNA-seq enables comprehensive analysis of the transcriptome for various purposes. However, this technology generally generates massive amounts of sequencing reads with a shorter read length. Consequently, fast, accurate, and flexible tools are needed for assembling raw RNA-seq data into full-length transcripts and quantifying their expression levels. In this protocol, we report TransBorrow, a novel transcriptome assembly software specifically designed for short RNA-seq reads. TransBorrow is employed in conjunction with a splice-aware alignment tool (e.g. Hisat2 and Star) and some other transcriptome assembly tools (e.g. StringTie, Cufflinks, and Scallop). The protocol encompasses all necessary steps, starting from downloading and processing raw sequencing data to assembling the full-length transcripts and quantifying their expressed abundances. The execution time of the protocol may vary depending on the sizes of processed datasets and computational platforms.
Keywords: RNA-seq data; splice variants; transcriptome assembly.
© The Author(s) 2023. Published by Oxford University Press.
Figures



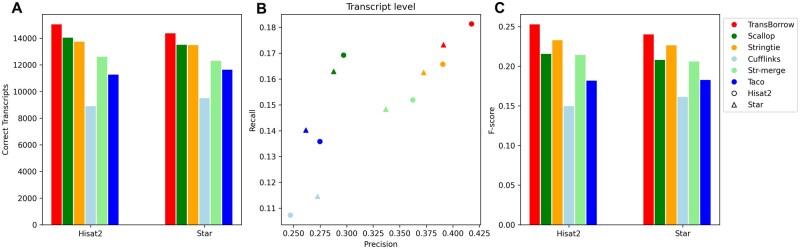
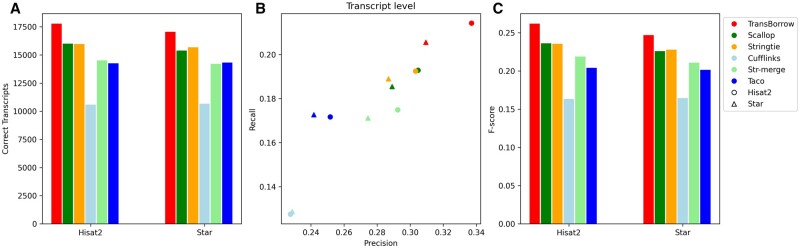
Similar articles
-
StringTie enables improved reconstruction of a transcriptome from RNA-seq reads.Nat Biotechnol. 2015 Mar;33(3):290-5. doi: 10.1038/nbt.3122. Epub 2015 Feb 18. Nat Biotechnol. 2015. PMID: 25690850 Free PMC article.
-
Improved transcriptome assembly using a hybrid of long and short reads with StringTie.PLoS Comput Biol. 2022 Jun 1;18(6):e1009730. doi: 10.1371/journal.pcbi.1009730. eCollection 2022 Jun. PLoS Comput Biol. 2022. PMID: 35648784 Free PMC article.
-
Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown.Nat Protoc. 2016 Sep;11(9):1650-67. doi: 10.1038/nprot.2016.095. Epub 2016 Aug 11. Nat Protoc. 2016. PMID: 27560171 Free PMC article.
-
Mapping RNA-seq reads to transcriptomes efficiently based on learning to hash method.Comput Biol Med. 2020 Jan;116:103539. doi: 10.1016/j.compbiomed.2019.103539. Epub 2019 Nov 13. Comput Biol Med. 2020. PMID: 31765913 Review.
-
RNA-seq data science: From raw data to effective interpretation.Front Genet. 2023 Mar 13;14:997383. doi: 10.3389/fgene.2023.997383. eCollection 2023. Front Genet. 2023. PMID: 36999049 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources