

Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
- PMID: 38017433
- PMCID: PMC10683363
- DOI: 10.1186/s12943-023-01873-0
Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
Abstract
The advent of iPSCs has brought about a significant transformation in stem cell research, opening up promising avenues for advancing cancer treatment. The formation of cancer is a multifaceted process influenced by genetic, epigenetic, and environmental factors. iPSCs offer a distinctive platform for investigating the origin of cancer, paving the way for novel approaches to cancer treatment, drug testing, and tailored medical interventions. This review article will provide an overview of the science behind iPSCs, the current limitations and challenges in iPSC-based cancer therapy, the ethical and social implications, and the comparative analysis with other stem cell types for cancer treatment. The article will also discuss the applications of iPSCs in tumorigenesis, the future of iPSCs in tumorigenesis research, and highlight successful case studies utilizing iPSCs in tumorigenesis research. The conclusion will summarize the advancements made in iPSC-based tumorigenesis research and the importance of continued investment in iPSC research to unlock the full potential of these cells.
Keywords: Immunotherapies; Induced pluripotent stem cells; Personalized medicine; Regenerative medicine; Therapy; Tumorigenesis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

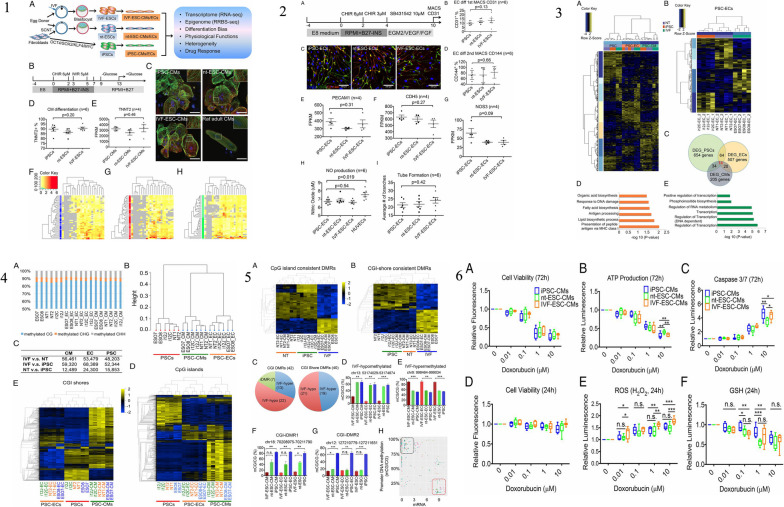
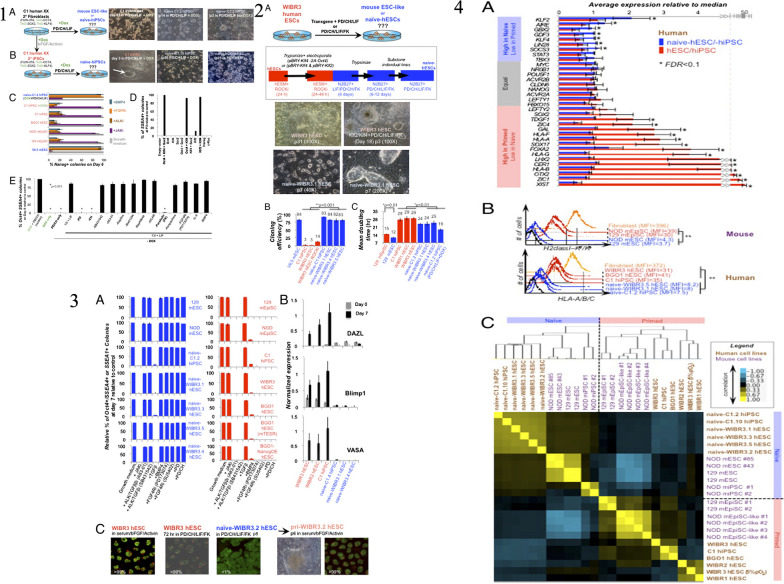
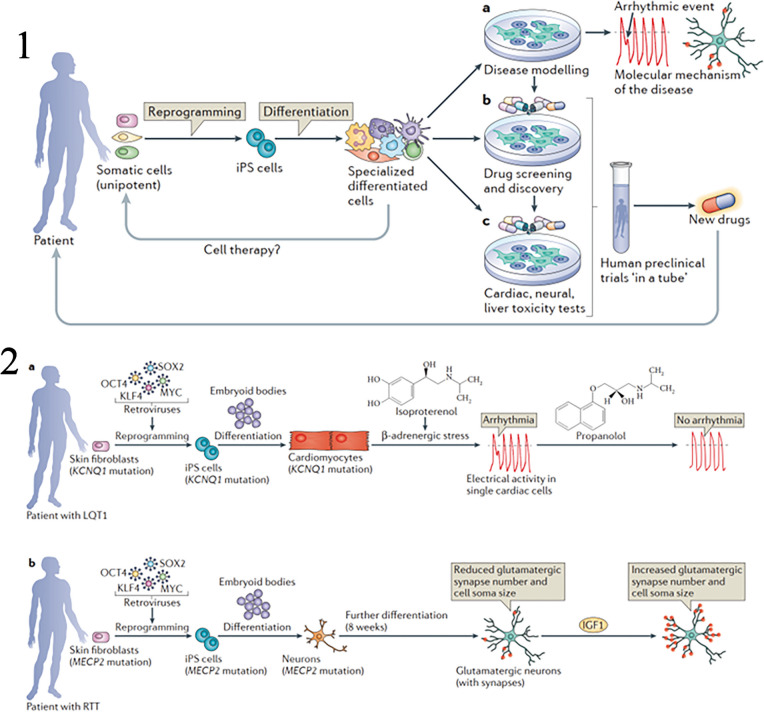
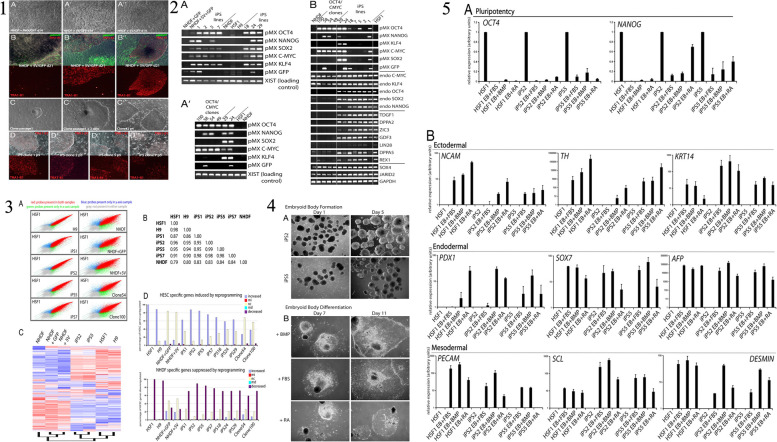

Similar articles
-
Exploiting urine-derived induced pluripotent stem cells for advancing precision medicine in cell therapy, disease modeling, and drug testing.J Biomed Sci. 2024 May 9;31(1):47. doi: 10.1186/s12929-024-01035-4. J Biomed Sci. 2024. PMID: 38724973 Free PMC article. Review.
-
Induced pluripotent stem cell consensus genes: implication for the risk of tumorigenesis and cancers in induced pluripotent stem cell therapy.Stem Cells Dev. 2012 Apr 10;21(6):955-64. doi: 10.1089/scd.2011.0649. Epub 2012 Feb 15. Stem Cells Dev. 2012. PMID: 22185567
-
Cancer cells as a new source of induced pluripotent stem cells.Stem Cell Res Ther. 2022 Sep 5;13(1):459. doi: 10.1186/s13287-022-03145-y. Stem Cell Res Ther. 2022. PMID: 36064437 Free PMC article. Review.
-
Induced pluripotency and oncogenic transformation are related processes.Stem Cells Dev. 2013 Jan 1;22(1):37-50. doi: 10.1089/scd.2012.0375. Epub 2012 Oct 26. Stem Cells Dev. 2013. PMID: 22998387 Free PMC article.
-
Possible Strategies to Reduce the Tumorigenic Risk of Reprogrammed Normal and Cancer Cells.Int J Mol Sci. 2024 May 9;25(10):5177. doi: 10.3390/ijms25105177. Int J Mol Sci. 2024. PMID: 38791215 Free PMC article. Review.
Cited by
-
Neuronal Cell Differentiation of iPSCs for the Clinical Treatment of Neurological Diseases.Biomedicines. 2024 Jun 18;12(6):1350. doi: 10.3390/biomedicines12061350. Biomedicines. 2024. PMID: 38927557 Free PMC article. Review.
-
Macrophage variants in laboratory research: most are well done, but some are RAW.Front Cell Infect Microbiol. 2024 Oct 9;14:1457323. doi: 10.3389/fcimb.2024.1457323. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 39445217 Free PMC article. Review.
-
Stem Cells and Infertility: A Review of Clinical Applications and Legal Frameworks.J Pers Med. 2024 Jan 24;14(2):135. doi: 10.3390/jpm14020135. J Pers Med. 2024. PMID: 38392569 Free PMC article. Review.
-
Epigenetic and Immune Mechanisms Linking Breastfeeding to Lower Breast Cancer Rates.Med Sci Monit. 2024 Nov 5;30:e945451. doi: 10.12659/MSM.945451. Med Sci Monit. 2024. PMID: 39497379 Free PMC article. Review.
-
Synergistic potential of stem cells and microfluidics in regenerative medicine.Mol Cell Biochem. 2024 Sep 16. doi: 10.1007/s11010-024-05108-8. Online ahead of print. Mol Cell Biochem. 2024. PMID: 39285093 Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
