

Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis
- PMID: 38003380
- PMCID: PMC10671649
- DOI: 10.3390/ijms242216190
Structural and pKa Estimation of the Amphipathic HR1 in SARS-CoV-2: Insights from Constant pH MD, Linear vs. Nonlinear Normal Mode Analysis
Abstract
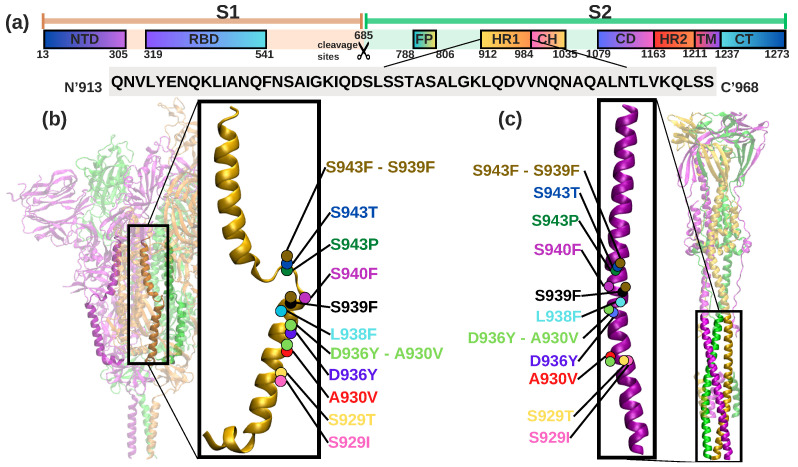
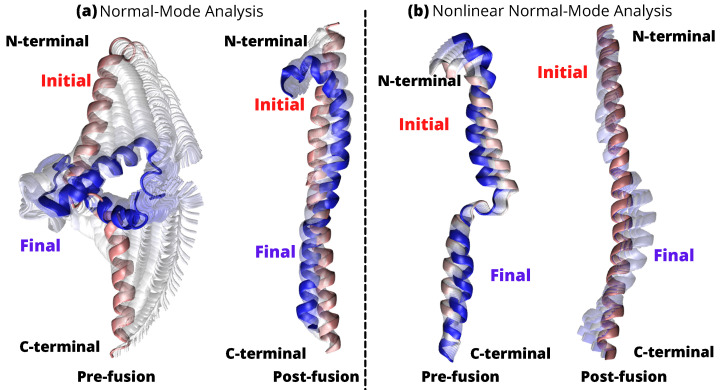
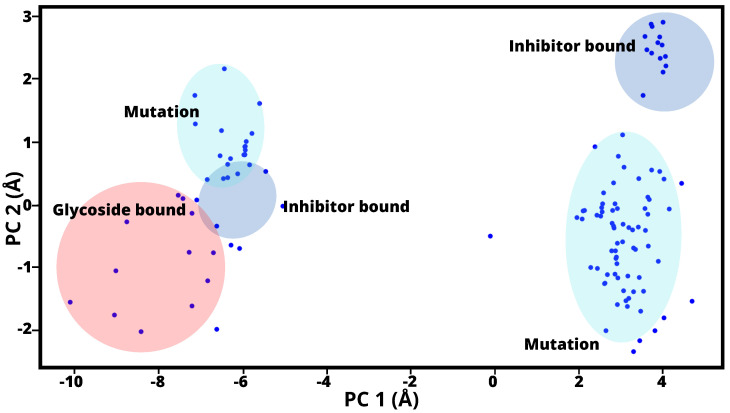
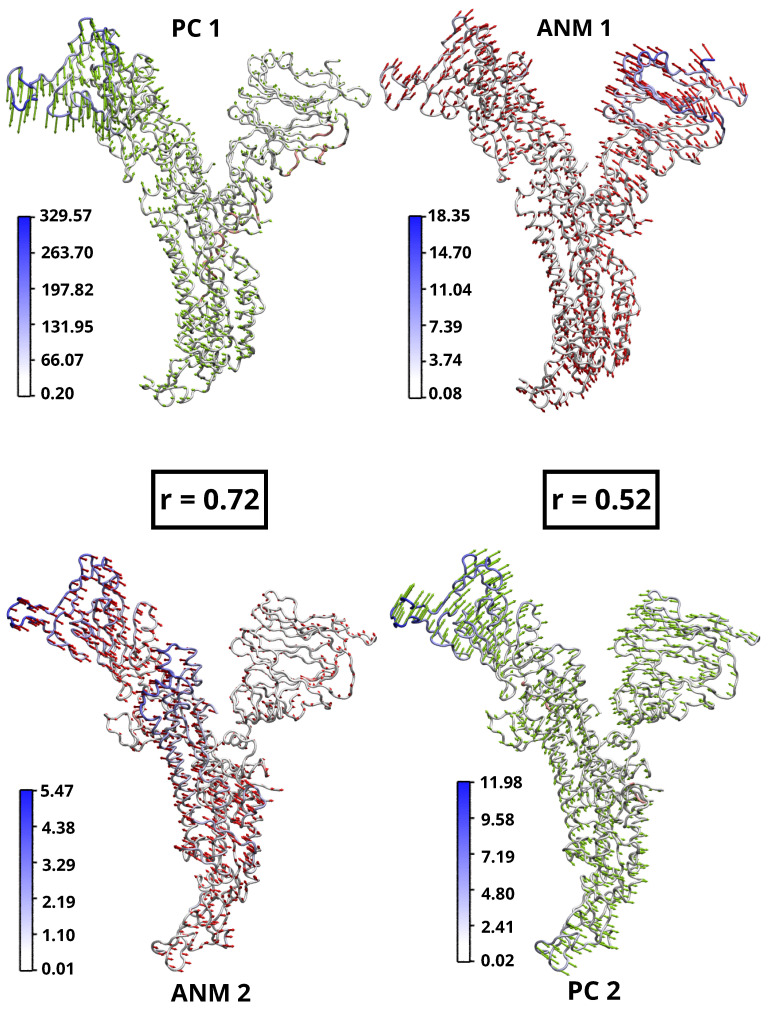
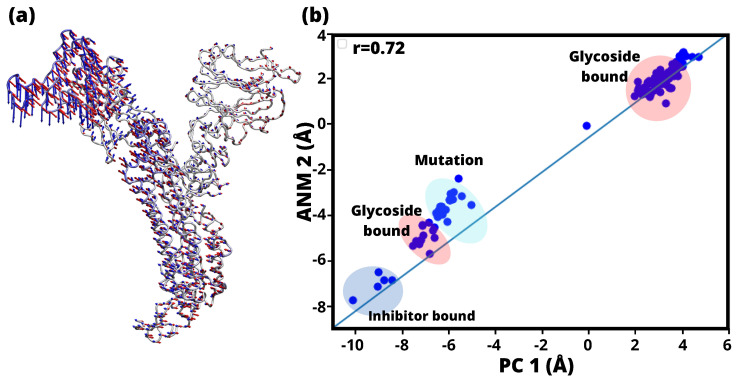
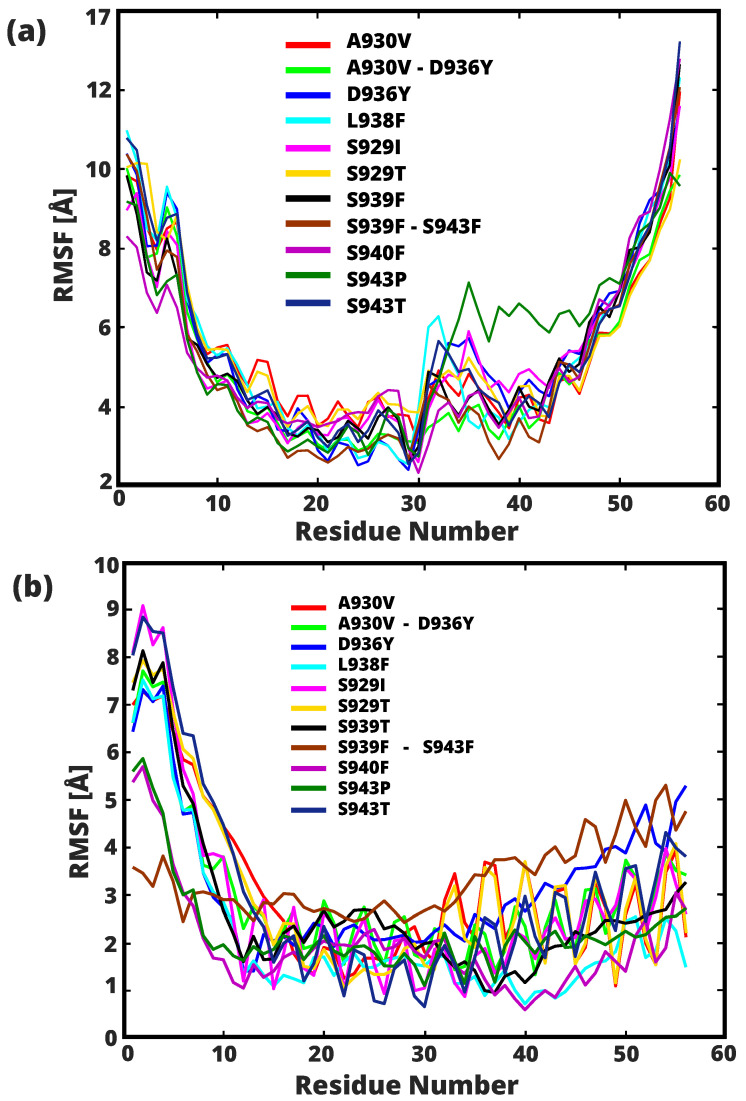
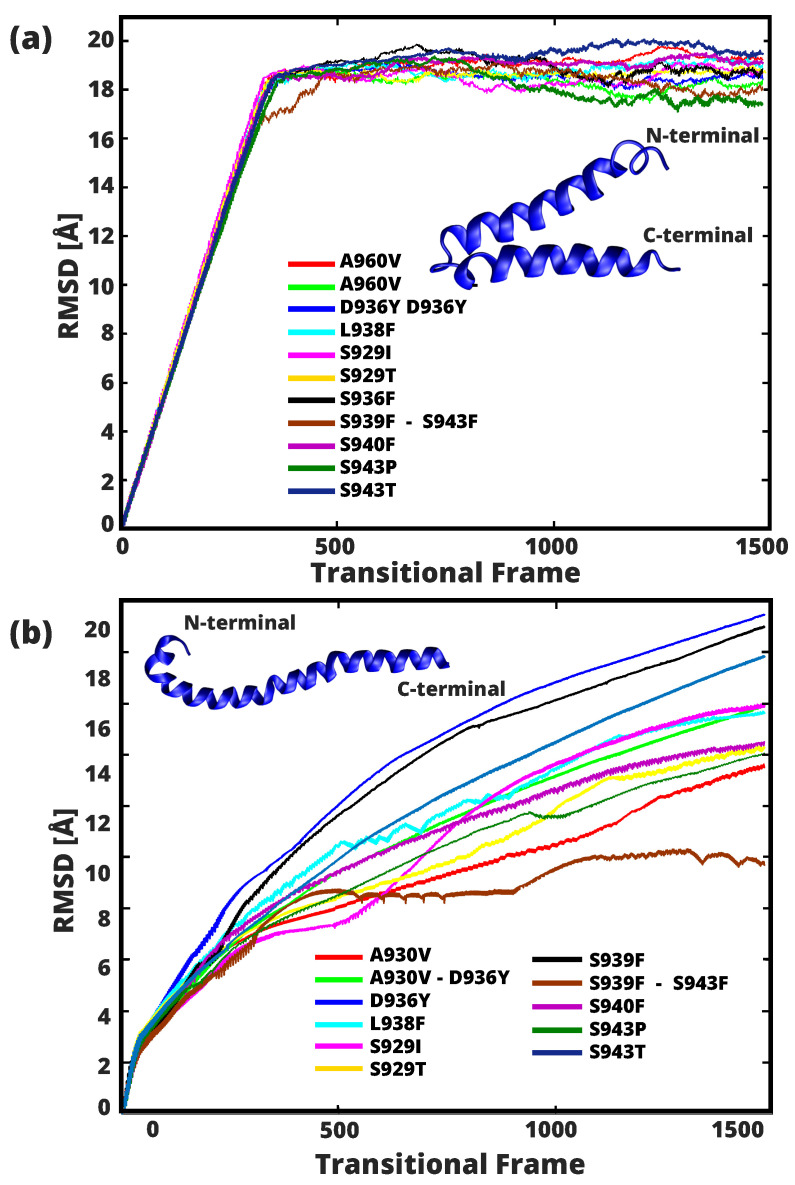
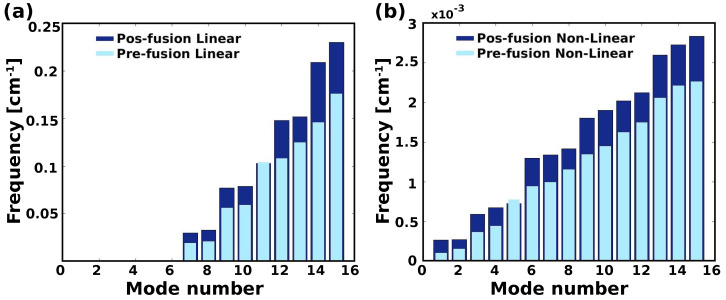
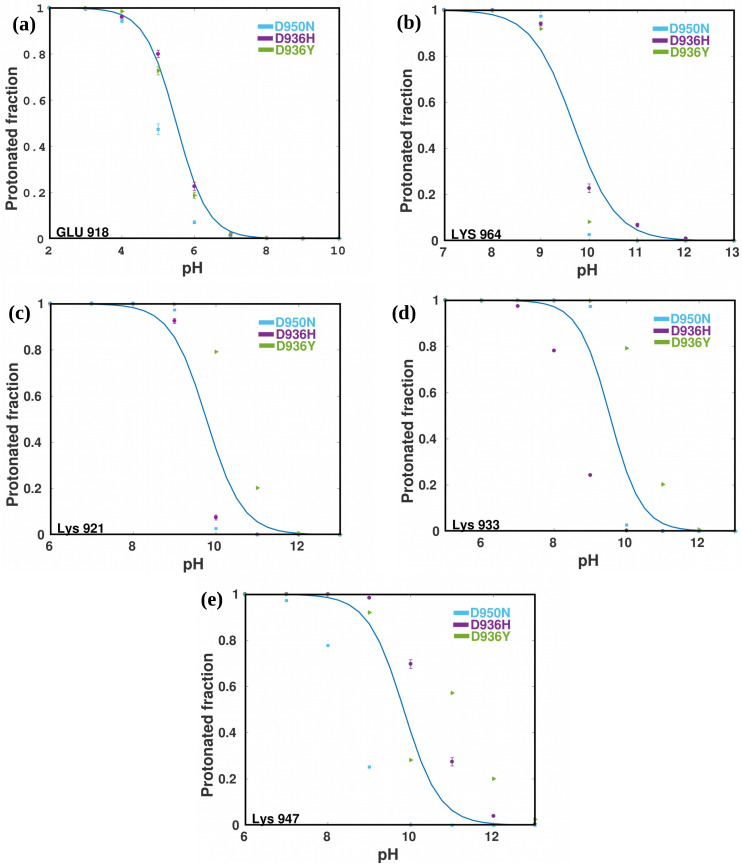
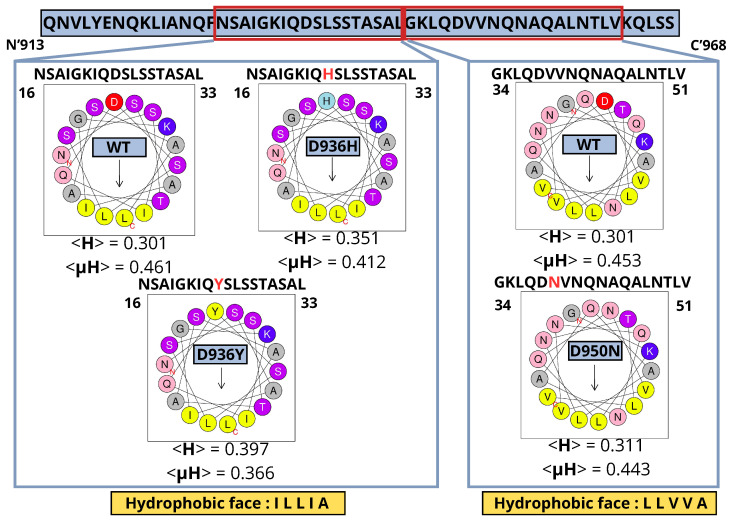
A comprehensive understanding of molecular interactions and functions is imperative for unraveling the intricacies of viral protein behavior and conformational dynamics during cellular entry. Focusing on the SARS-CoV-2 spike protein (SARS-CoV-2 sp), a Principal Component Analysis (PCA) on a subset comprising 131 A-chain structures in presence of various inhibitors was conducted. Our analyses unveiled a compelling correlation between PCA modes and Anisotropic Network Model (ANM) modes, underscoring the reliability and functional significance of low-frequency modes in adapting to diverse inhibitor binding scenarios. The role of HR1 in viral processing, both linear Normal Mode Analysis (NMA) and Nonlinear NMA were implemented. Linear NMA exhibited substantial inter-structure variability, as evident from a higher Root Mean Square Deviation (RMSD) range (7.30 Å), nonlinear NMA show stability throughout the simulations (RMSD 4.85 Å). Frequency analysis further emphasized that the energy requirements for conformational changes in nonlinear modes are notably lower compared to their linear counterparts. Using simulations of molecular dynamics at constant pH (cpH-MD), we successfully predicted the pKa order of the interconnected residues within the HR1 mutations at lower pH values, suggesting a transition to a post-fusion structure. The pKa determination study illustrates the profound effects of pH variations on protein structure. Key results include pKa values of 9.5179 for lys-921 in the D936H mutant, 9.50 for the D950N mutant, and a slightly higher value of 10.49 for the D936Y variant. To further understand the behavior and physicochemical characteristics of the protein in a biologically relevant setting, we also examine hydrophobic regions in the prefused states of the HR1 protein mutants D950N, D936Y, and D936H in our study. This analysis was conducted to ascertain the hydrophobic moment of the protein within a lipid environment, shedding light on its behavior and physicochemical properties in a biologically relevant context.
Keywords: HR1; SARS-CoV-2; constant pH molecular dynamics; mutations; nonlinear and linear—Normal Mode Analysis; principal component analysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect.Molecules. 2021 Apr 30;26(9):2622. doi: 10.3390/molecules26092622. Molecules. 2021. PMID: 33946306 Free PMC article.
-
Insights into the structural and dynamical changes of spike glycoprotein mutations associated with SARS-CoV-2 host receptor binding.J Biomol Struct Dyn. 2022 Jan;40(1):263-275. doi: 10.1080/07391102.2020.1811774. Epub 2020 Aug 27. J Biomol Struct Dyn. 2022. PMID: 32851910 Free PMC article.
-
Repurposing fusion inhibitor peptide against SARS-CoV-2.J Comput Chem. 2021 Dec 15;42(32):2283-2293. doi: 10.1002/jcc.26758. Epub 2021 Sep 30. J Comput Chem. 2021. PMID: 34591335
-
How helpful were molecular dynamics simulations in shaping our understanding of SARS-CoV-2 spike protein dynamics?Int J Biol Macromol. 2023 Jul 1;242(Pt 4):125153. doi: 10.1016/j.ijbiomac.2023.125153. Epub 2023 Jun 1. Int J Biol Macromol. 2023. PMID: 37268078 Free PMC article. Review.
-
Molecular Dynamics Studies on the Structural Characteristics for the Stability Prediction of SARS-CoV-2.Int J Mol Sci. 2021 Aug 13;22(16):8714. doi: 10.3390/ijms22168714. Int J Mol Sci. 2021. PMID: 34445414 Free PMC article. Review.
References
-
- Zimmerman M.I., Porter J.R., Ward M.D., Singh S., Vithani N., Meller A., Mallimadugula U.L., Kuhn C.E., Borowsky J.H., Wiewiora R.P., et al. SARS-CoV-2 Simulations Go Exascale to Predict Dramatic Spike Opening and Cryptic Pockets across the Proteome. Nat. Chem. 2021;13:651–659. doi: 10.1038/s41557-021-00707-0. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
