

Leveraging the Fragment Molecular Orbital Method to Explore the PLK1 Kinase Binding Site and Polo-Box Domain for Potent Small-Molecule Drug Design
- PMID: 37958623
- PMCID: PMC10650754
- DOI: 10.3390/ijms242115639
Leveraging the Fragment Molecular Orbital Method to Explore the PLK1 Kinase Binding Site and Polo-Box Domain for Potent Small-Molecule Drug Design
Abstract
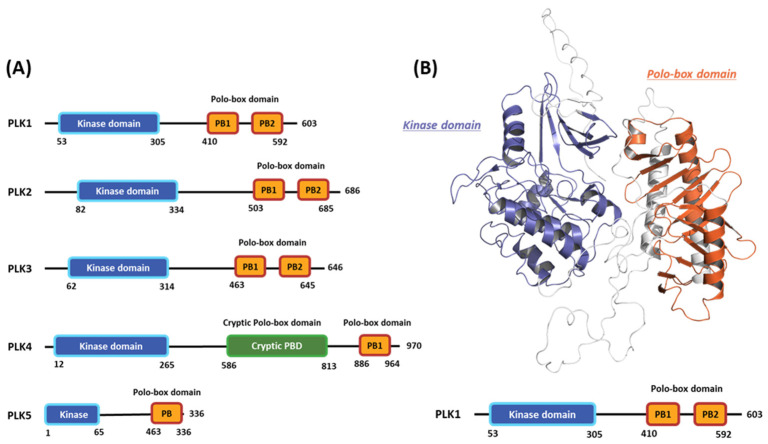
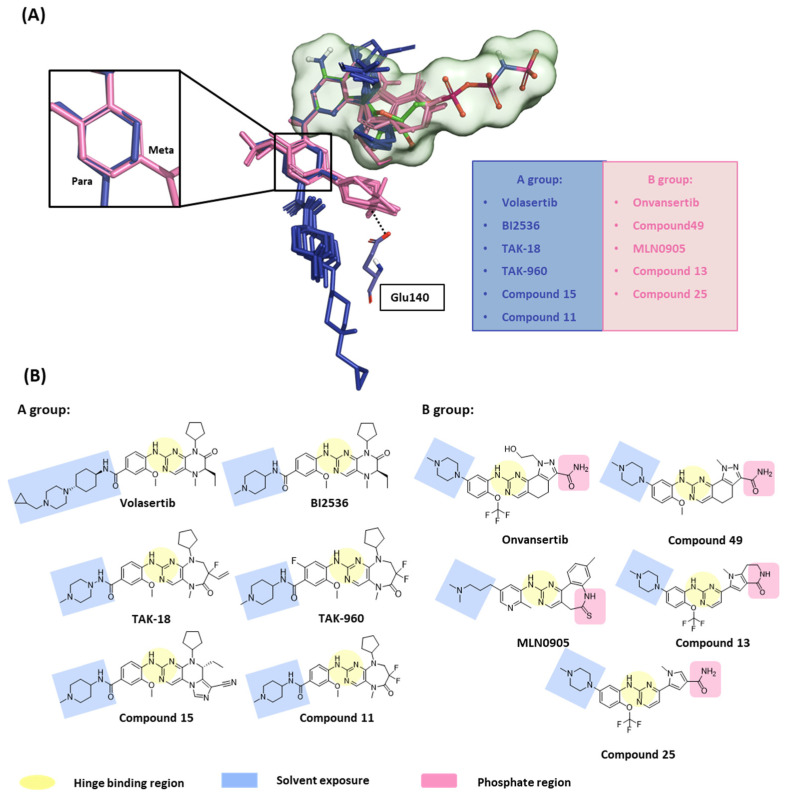
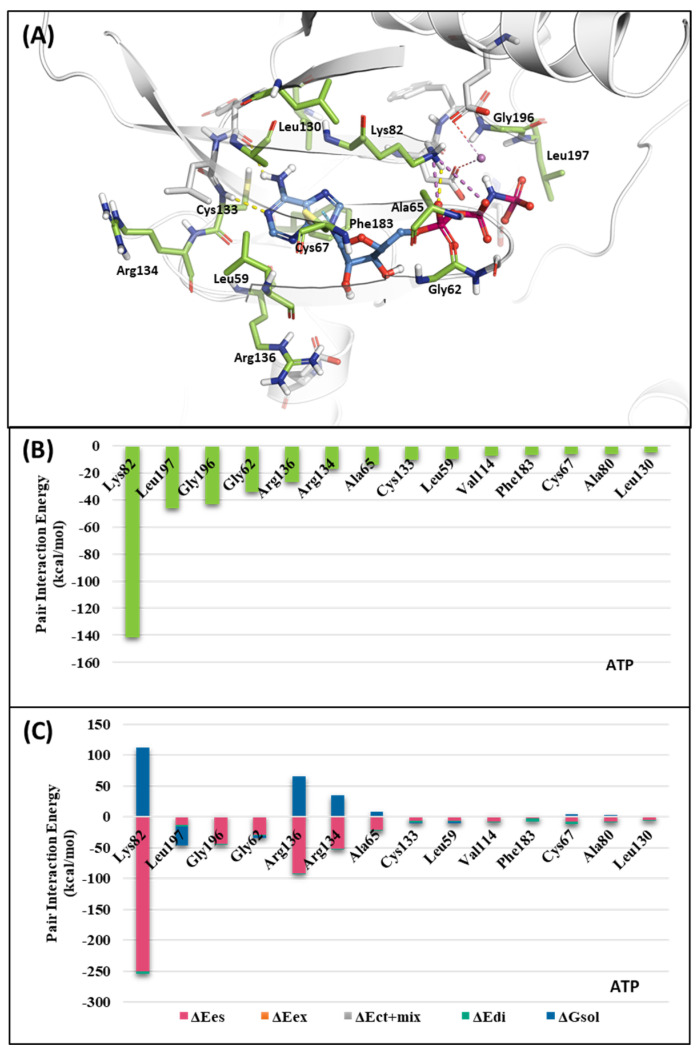
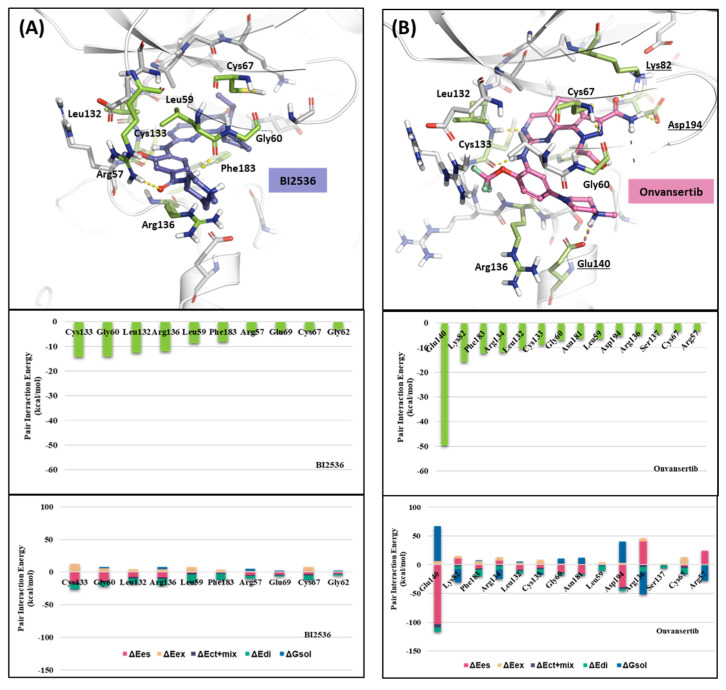
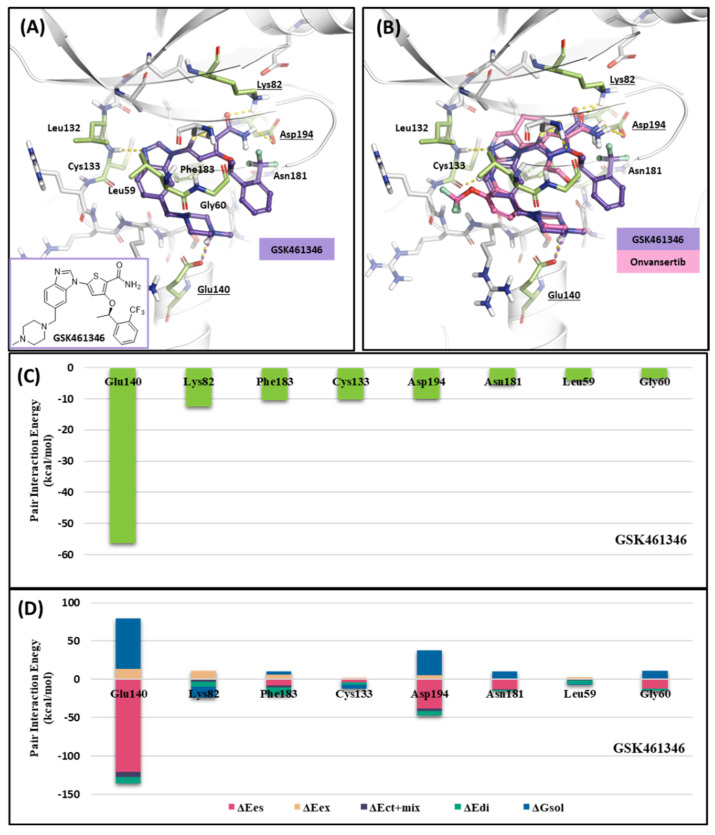
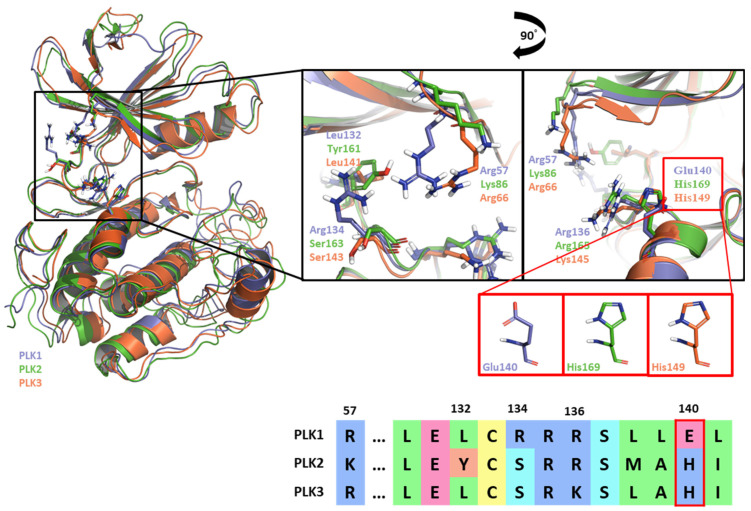
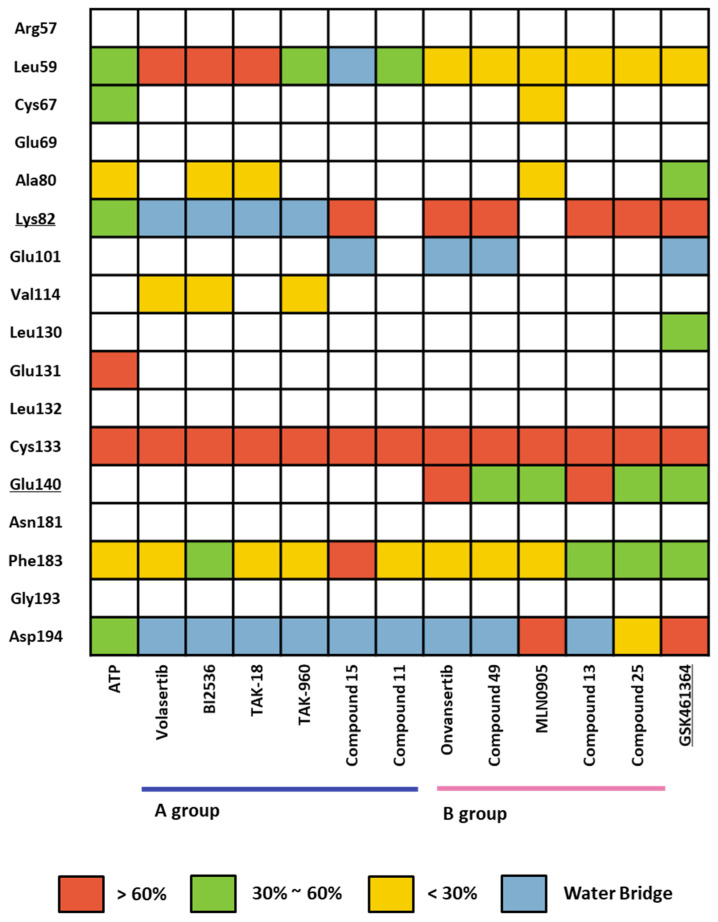
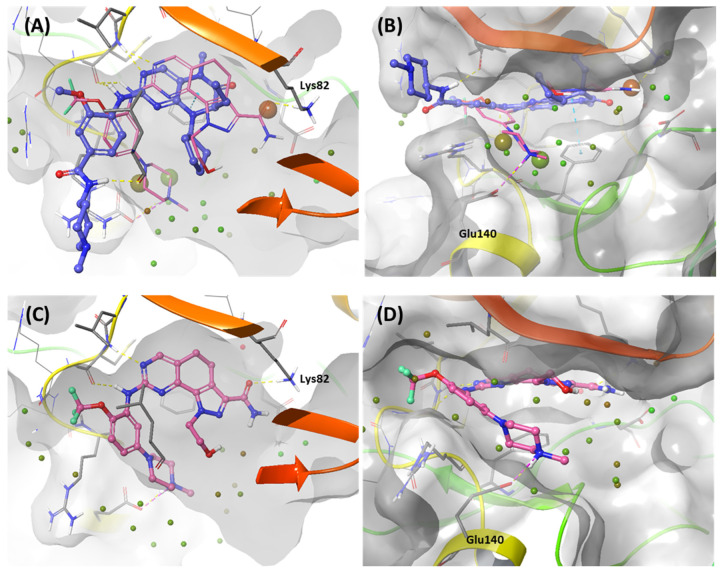
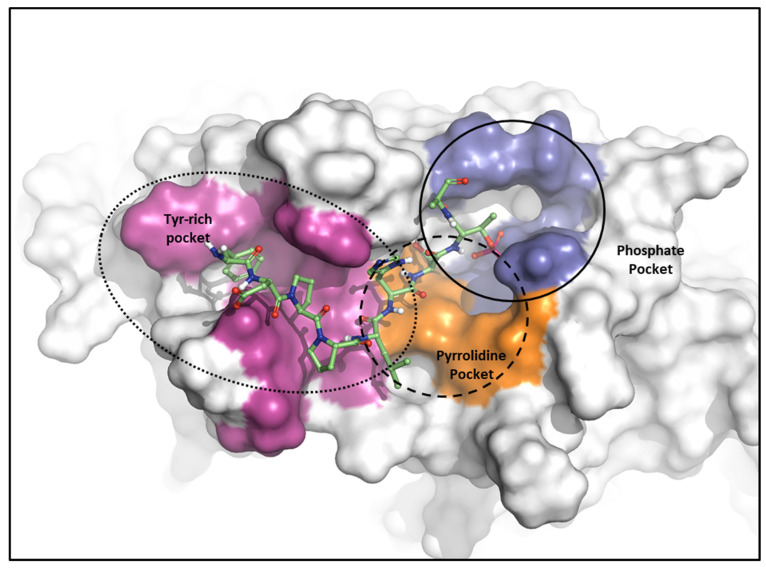
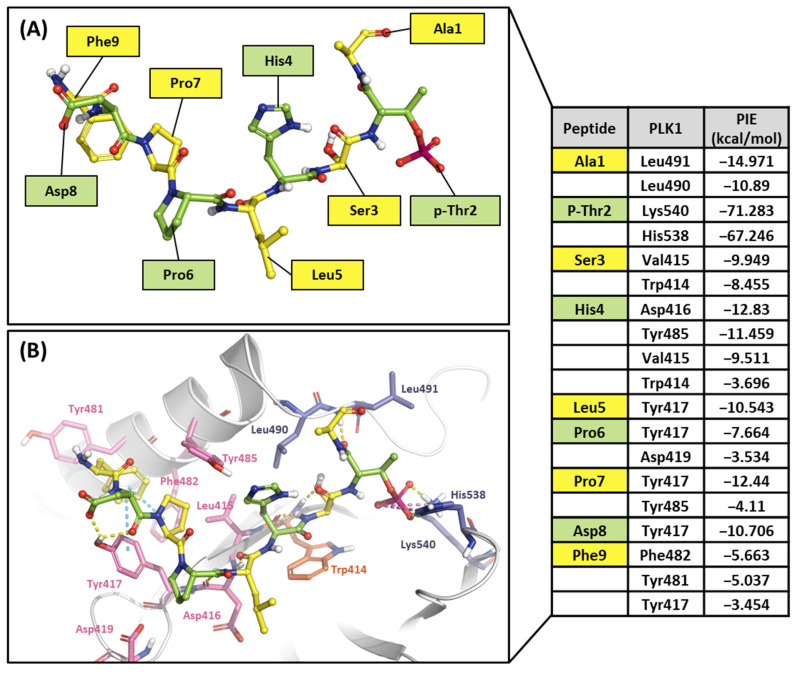
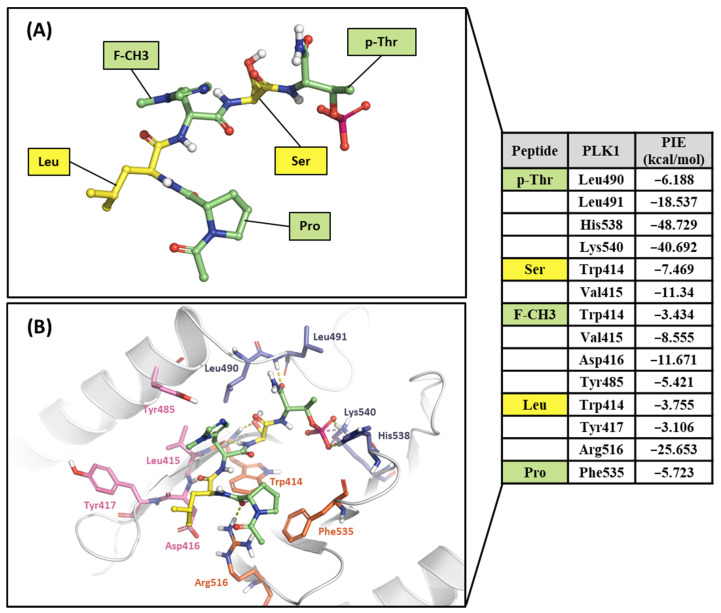
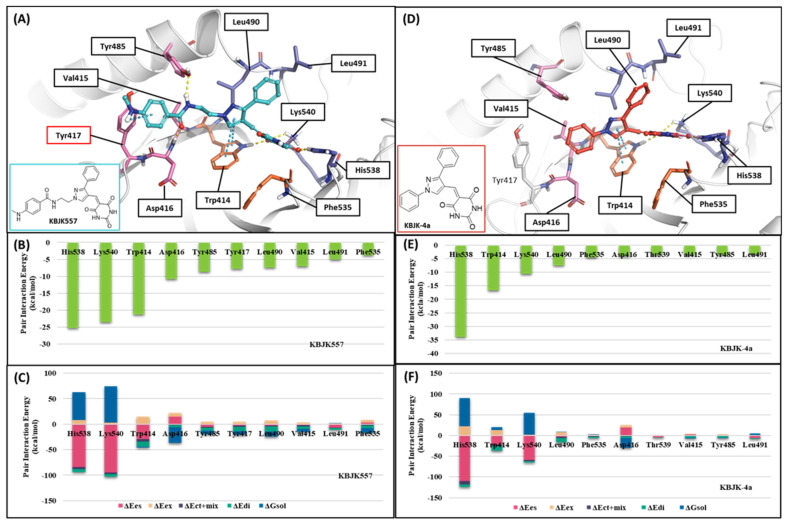
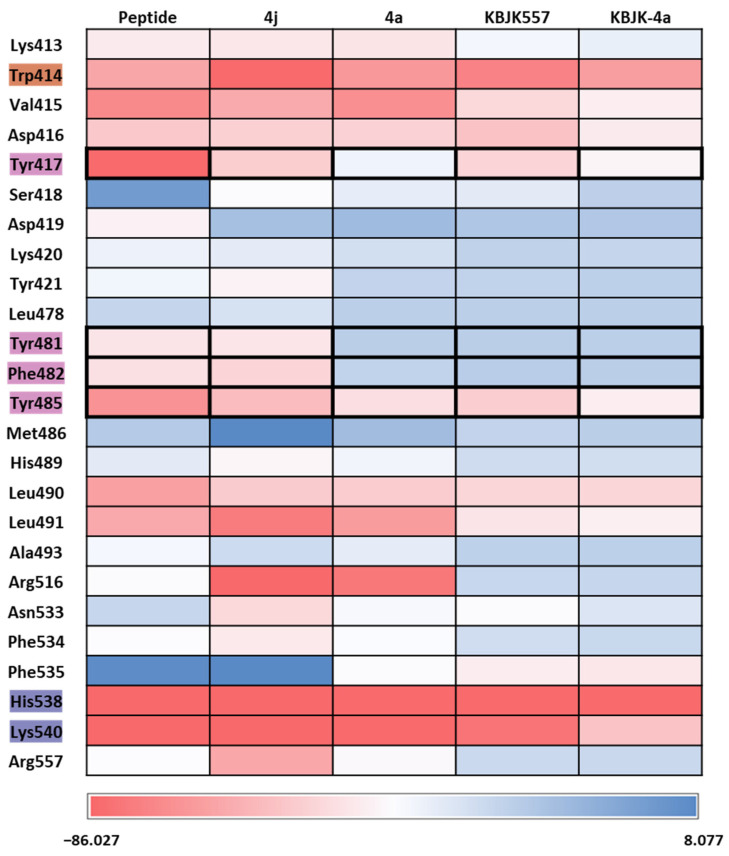
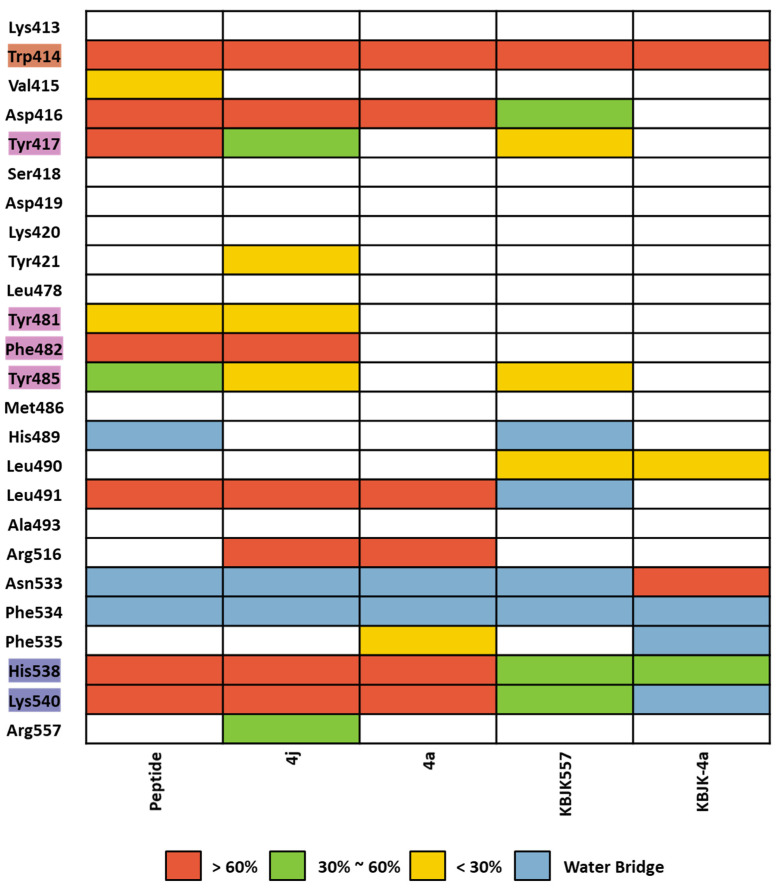
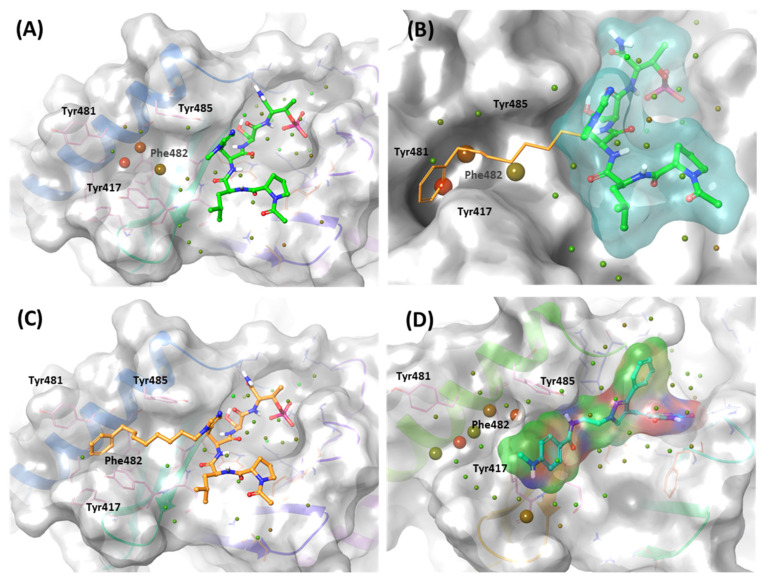
Polo-like kinase 1 (PLK1) plays a pivotal role in cell division regulation and emerges as a promising therapeutic target for cancer treatment. Consequently, the development of small-molecule inhibitors targeting PLK1 has become a focal point in contemporary research. The adenosine triphosphate (ATP)-binding site and the polo-box domain in PLK1 present crucial interaction sites for these inhibitors, aiming to disrupt the protein's function. However, designing potent and selective small-molecule inhibitors can be challenging, requiring a deep understanding of protein-ligand interaction mechanisms at these binding sites. In this context, our study leverages the fragment molecular orbital (FMO) method to explore these site-specific interactions in depth. Using the FMO approach, we used the FMO method to elucidate the molecular mechanisms of small-molecule drugs binding to these sites to design PLK1 inhibitors that are both potent and selective. Our investigation further entailed a comparative analysis of various PLK1 inhibitors, each characterized by distinct structural attributes, helping us gain a better understanding of the relationship between molecular structure and biological activity. The FMO method was particularly effective in identifying key binding features and predicting binding modes for small-molecule ligands. Our research also highlighted specific "hot spot" residues that played a critical role in the selective and robust binding of PLK1. These findings provide valuable insights that can be used to design new and effective PLK1 inhibitors, which can have significant implications for developing anticancer therapeutics.
Keywords: fragment molecular orbital method; molecular dynamics simulation; polo-like kinase 1; protein-protein interaction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
In silico identification of putative bifunctional Plk1 inhibitors by integrative virtual screening and structural dynamics approach.J Theor Biol. 2016 Jan 7;388:72-84. doi: 10.1016/j.jtbi.2015.10.006. Epub 2015 Oct 19. J Theor Biol. 2016. PMID: 26493360
-
Design and synthesis of a cell-permeable, drug-like small molecule inhibitor targeting the polo-box domain of polo-like kinase 1.PLoS One. 2014 Sep 11;9(9):e107432. doi: 10.1371/journal.pone.0107432. eCollection 2014. PLoS One. 2014. PMID: 25211362 Free PMC article.
-
Structure-activity and mechanistic studies of non-peptidic inhibitors of the PLK1 polo box domain identified through REPLACE.Eur J Med Chem. 2022 Jan 5;227:113926. doi: 10.1016/j.ejmech.2021.113926. Epub 2021 Oct 21. Eur J Med Chem. 2022. PMID: 34735919 Free PMC article.
-
Molecular and enzoinformatics perspectives of targeting Polo-like kinase 1 in cancer therapy.Semin Cancer Biol. 2019 Jun;56:47-55. doi: 10.1016/j.semcancer.2017.11.004. Epub 2017 Nov 6. Semin Cancer Biol. 2019. PMID: 29122685 Review.
-
[Mining Polo-Box domain of Polo-like kinase 1 as a new therapeutic target for cancer].Sheng Wu Gong Cheng Xue Bao. 2020 Nov 25;36(11):2298-2312. doi: 10.13345/j.cjb.200132. Sheng Wu Gong Cheng Xue Bao. 2020. PMID: 33244925 Review. Chinese.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
