

TIPE proteins control directed migration of human T cells by directing GPCR and lipid second messenger signaling
- PMID: 37952106
- PMCID: PMC10890839
- DOI: 10.1093/jleuko/qiad141
TIPE proteins control directed migration of human T cells by directing GPCR and lipid second messenger signaling
Abstract
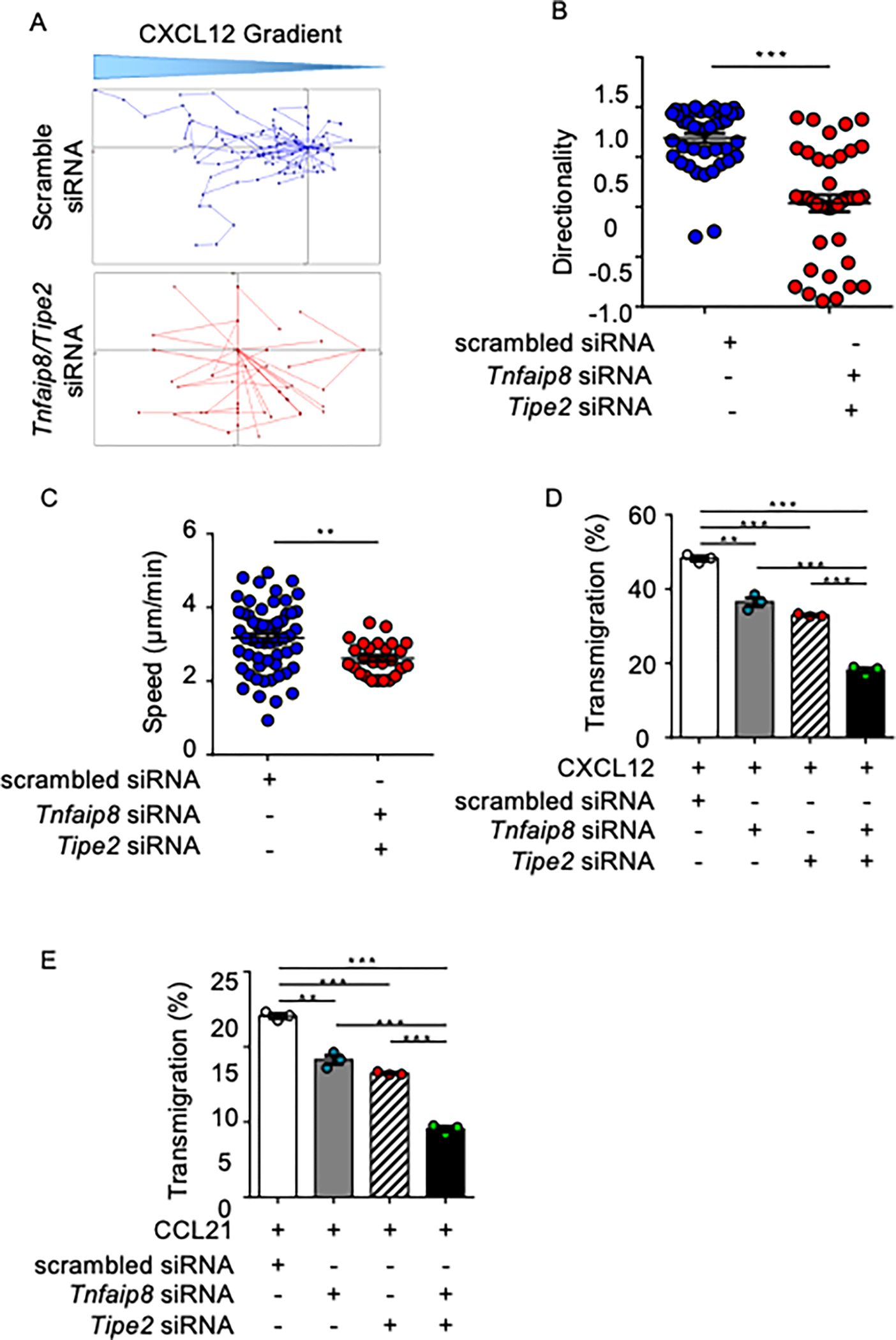
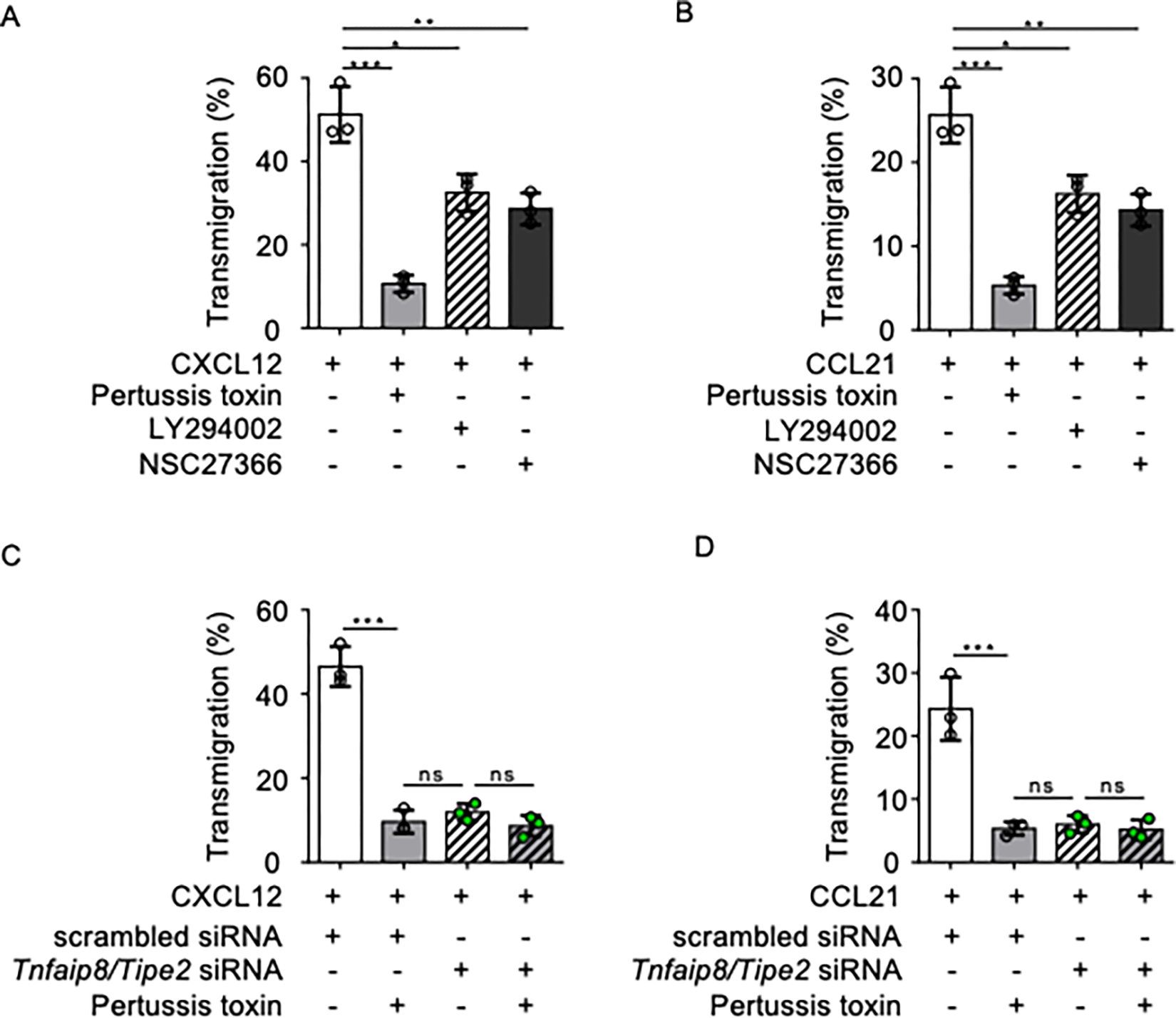
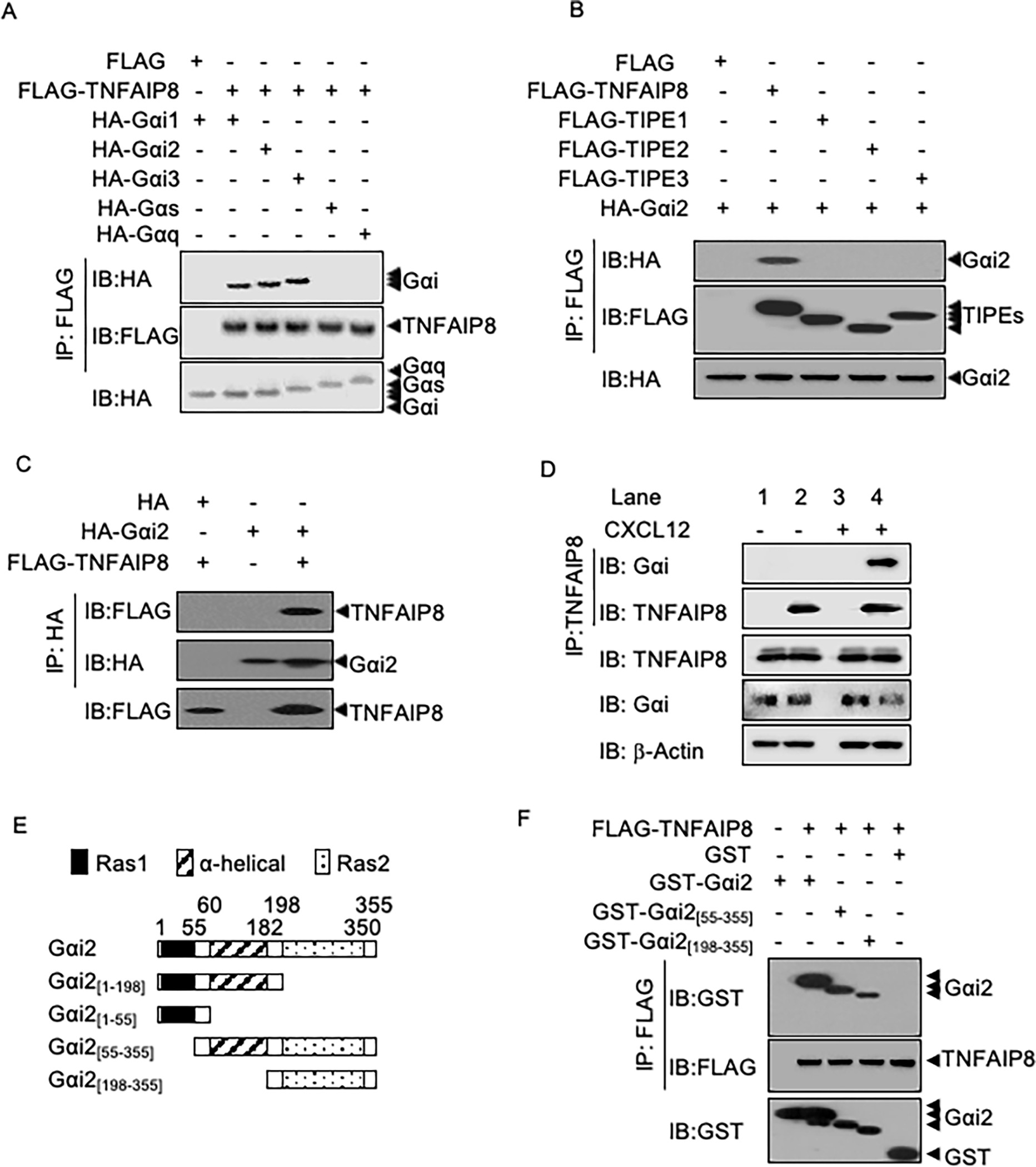
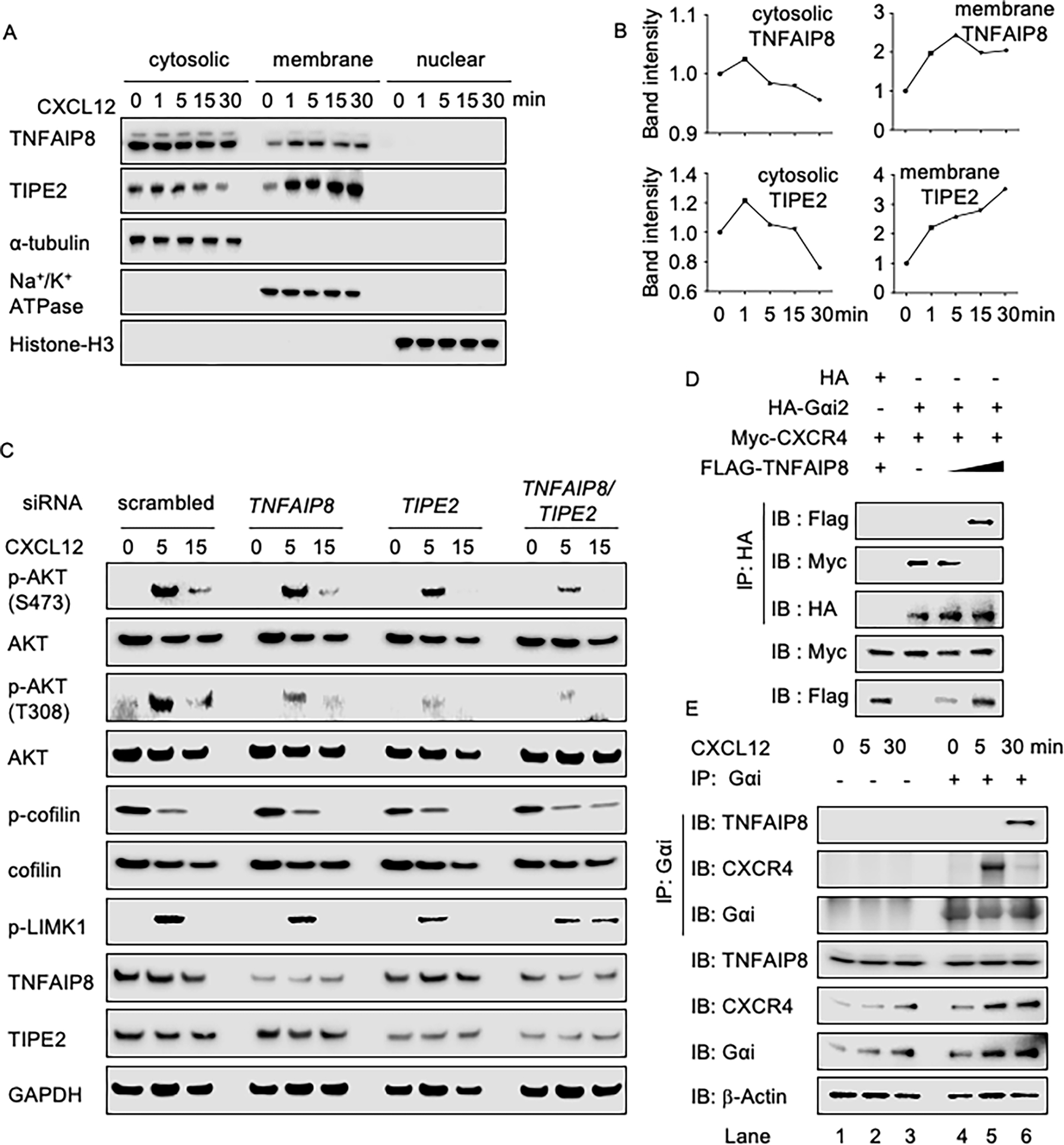
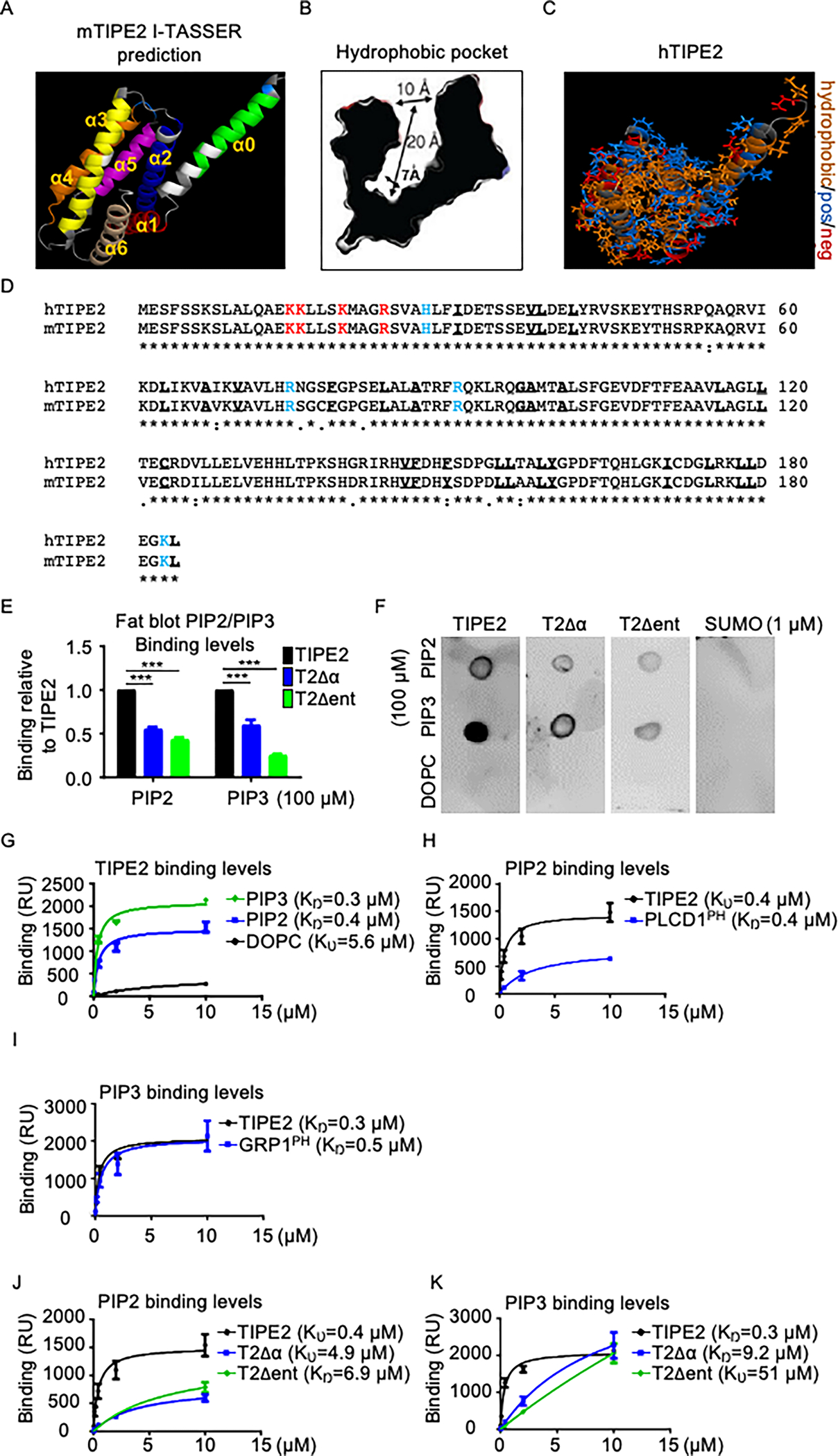
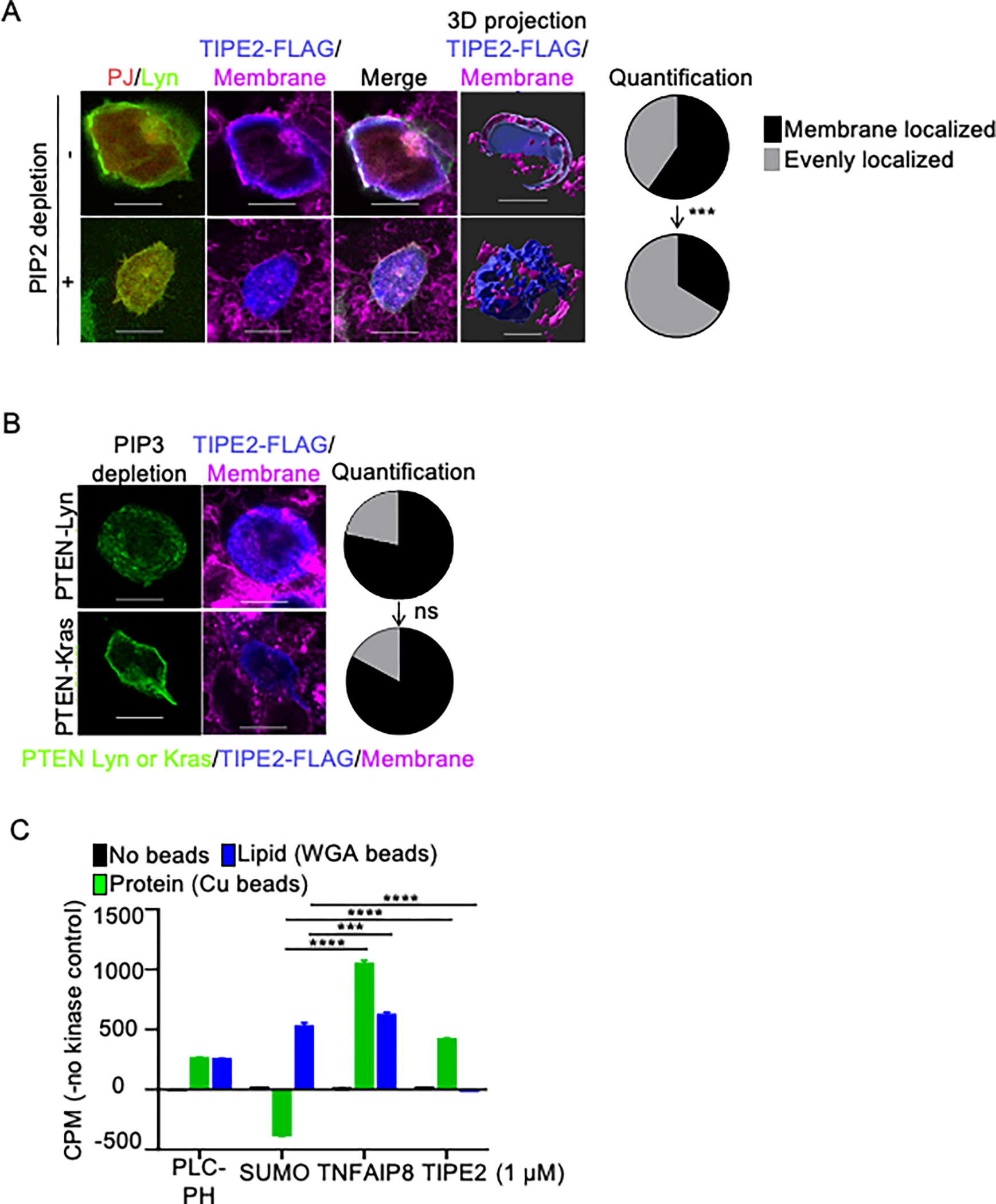
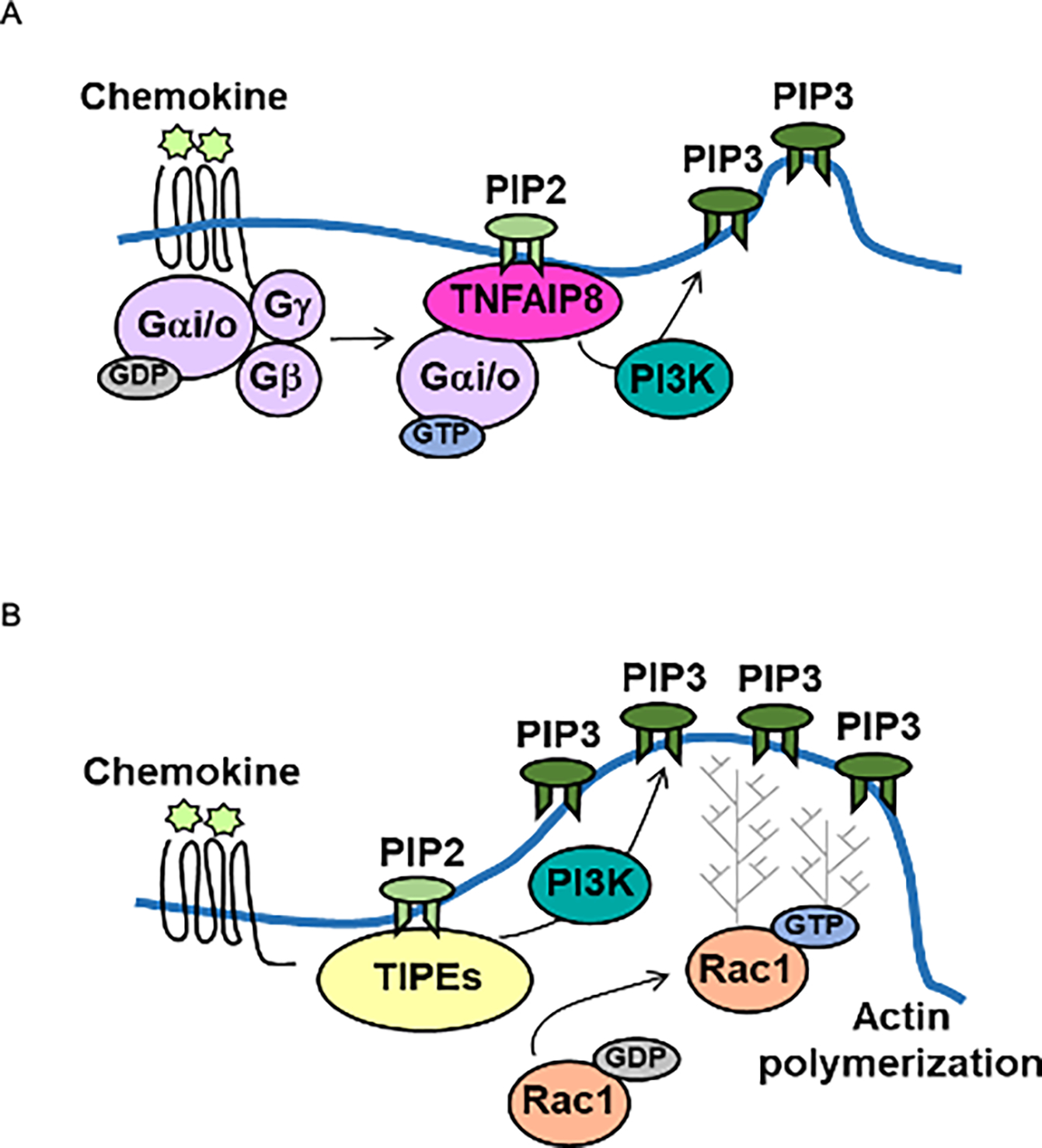
Tissue infiltration by circulating leukocytes via directed migration (also referred to as chemotaxis) is a common pathogenic mechanism of inflammatory diseases. G protein-coupled receptors (GPCRs) are essential for sensing chemokine gradients and directing the movement of leukocytes during immune responses. The tumor necrosis factor α-induced protein 8-like (TIPE or TNFAIP8L) family of proteins are newly described pilot proteins that control directed migration of murine leukocytes. However, how leukocytes integrate site-specific directional cues, such as chemokine gradients, and utilize GPCR and TIPE proteins to make directional decisions are not well understood. Using both gene knockdown and biochemical methods, we demonstrated here that 2 human TIPE family members, TNFAIP8 and TIPE2, were essential for directed migration of human CD4+ T cells. T cells deficient in both of these proteins completely lost their directionality. TNFAIP8 interacted with the Gαi subunit of heterotrimeric (α, β, γ) G proteins, whereas TIPE2 bound to PIP2 and PIP3 to spatiotemporally control immune cell migration. Using deletion and site-directed mutagenesis, we established that Gαi interacted with TNFAIP8 through its C-terminal amino acids, and that TIPE2 protein interacted with PIP2 and PIP3 through its positively charged amino acids on the α0 helix and at the grip-like entrance. We also discovered that TIPE protein membrane translocation (i.e. crucial for sensing chemokine gradients) was dependent on PIP2. Collectively, our work describes a new mechanistic paradigm for how human T cells integrate GPCR and phospholipid signaling pathways to control directed migration. These findings have implications for therapeutically targeting TIPE proteins in human inflammatory and autoimmune diseases.
Keywords: PI3K signaling; TIPE proteins; chemoattractant sensing; directed migration; heterotrimeric g proteins.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Leukocyte Biology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
TIPE polarity proteins are required for mucosal deployment of T lymphocytes and mucosal defense against bacterial infection.Mol Biomed. 2021 Dec 23;2(1):41. doi: 10.1186/s43556-021-00059-8. Mol Biomed. 2021. PMID: 34939151 Free PMC article.
-
Tumor necrosis factor-α induced protein 8 (TNFAIP8/TIPE) family is differentially expressed in oral cancer and regulates tumorigenesis through Akt/mTOR/STAT3 signaling cascade.Life Sci. 2021 Dec 15;287:120118. doi: 10.1016/j.lfs.2021.120118. Epub 2021 Nov 2. Life Sci. 2021. PMID: 34740574
-
Regulation of inflammation and tumorigenesis by the TIPE family of phospholipid transfer proteins.Cell Mol Immunol. 2017 Jun;14(6):482-487. doi: 10.1038/cmi.2017.4. Epub 2017 Mar 13. Cell Mol Immunol. 2017. PMID: 28287114 Free PMC article. Review.
-
The TIPE Molecular Pilot That Directs Lymphocyte Migration in Health and Inflammation.Sci Rep. 2020 Apr 20;10(1):6617. doi: 10.1038/s41598-020-63629-w. Sci Rep. 2020. PMID: 32313148 Free PMC article.
-
TIPE Family of Proteins and Its Implications in Different Chronic Diseases.Int J Mol Sci. 2018 Sep 29;19(10):2974. doi: 10.3390/ijms19102974. Int J Mol Sci. 2018. PMID: 30274259 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
